Nous présentons le cas d’un homme de 43 ans qui est un ex-fumeur. Il a présenté une dyspnée et une hypoxie progressives, et la radiographie pulmonaire a montré des altérations interstitielles diffuses bilatérales. Le scanner thoracique a révélé des opacités en verre dépoli bilatérales aggravées, sans signe de consolidation focale ou d’altérations interstitielles (Fig. 1a). Le patient a été intubé et ventilé et une biopsie pulmonaire a été réalisée. La biopsie a montré une combinaison d’infiltrat lymphocytaire interstitiel avec une architecture altérée dans de nombreuses zones, associée à des éléments de bronchiolite oblitérante qui touchaient principalement les voies aériennes au niveau des canaux alvéolaires, mais dans quelques zones, elle touchait également les bronchioles respiratoires. Cette combinaison est décrite comme une bronchiolite interstitielle (BIP) (Fig. 1b). Le patient s’est amélioré lentement, sans traitement spécifique. Lors de la consultation de suivi six mois plus tard, on a constaté une récupération complète. Un nouveau CT thoracique a montré une résolution complète de la PIF.
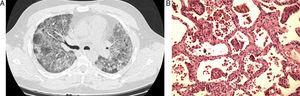
(A) Infiltrat interstitiel diffus dans les deux poumons, tel qu’observé sur le scanner thoracique. (B) Inflammation interstitielle et fibrose sous fort grossissement dans la pneumopathie interstitielle de bronchiolite.
La classification des pneumopathies interstitielles est largement basée sur les schémas observés dans les échantillons de biopsies ouvertes ou transbronchiques. En 2008, Mark et al. ont décrit une cohorte de 31 patients qui, après examen anatomopathologique, présentaient une pneumopathie interstitielle avec bronchiolite.1 100% des cas rapportés dans la seule série publiée présentaient des signes de bronchiolite oblitérante ainsi qu’une fibrose interstitielle. Il faut noter que la fibrose était à distance de la maladie bronchiolaire, ce qui suggère que la fibrose et la maladie bronchiolaire ne sont pas une conséquence directe l’une de l’autre. Dans tous les cas, la fibrose interstitielle a été observée à un taux plus élevé que l’inflammation interstitielle lymphocytaire.
La pneumopathie interstitielle a été définie comme un infiltrat interstitiel de lymphocytes et de fibrose.1 L’inflammation et la fibrose étaient présentes dans tous les cas. La pneumonie interstitielle avec bronchiolite présentait une réponse aux corticostéroïdes inférieure à celle de la bronchiolite oblitérante organisée (BOOP), mais supérieure à celle de la pneumonie interstitielle habituelle (UIP) et de la pneumonie interstitielle non spécifique/fibrose (NSIP). Des foyers fibroblastiques ont été observés dans seulement 21% des cas.1
Un autre diagnostic différentiel important qui doit également être pris en compte dans ce cas est la pneumopathie interstitielle associée à la bronchiolite respiratoire (RB-ILD). La RB-ILD est souvent observée chez des patients fumeurs ou ex-fumeurs, bien que son apparition ait également été décrite chez des non-fumeurs.2 On a obtenu des preuves indiquant une accumulation de macrophages à pigmentation sombre dans les bronchioles respiratoires et dans l’espace aérien environnant2 associés à une infiltration sous-muqueuse et péribronchiolaire parsemée de lymphocytes et d’histiocytes. Une fibrose péribronchiolaire peut également être observée.3 Dans cette affection, les foyers fibroblastiques ne sont pas observés, ce qui la différencie des autres pneumonies interstitielles idiopathiques.3
Ce cas pose la question de l’utilité des biopsies pulmonaires ouvertes étant plus fréquemment réalisées4 et de la nécessité d’une analyse histologique plus sophistiquée. L’examen anatomopathologique est moins utile lorsqu’il est obtenu plus tard dans l’évolution de la maladie ou après la mise en place d’un traitement.5
Notre cas est une contribution à la quantité limitée de données publiées à ce jour sur cette entité, qui est rapportée très rarement. Il s’agit du premier cas rapporté de résolution spontanée du PIF. Il est très important d’être plus conscient de cette entité, car de nombreux cas peuvent ne pas être diagnostiqués ou peuvent actuellement être mal diagnostiqués.