InterPro contient trois entités principales : les protéines, les signatures (également appelées « méthodes » ou « modèles ») et les entrées. Les protéines dans UniProtKB sont également les entités protéiques centrales dans InterPro. Les informations concernant les signatures qui correspondent de manière significative à ces protéines sont calculées au fur et à mesure que les séquences sont publiées par UniProtKB et ces résultats sont mis à la disposition du public (voir ci-dessous). Les correspondances entre les signatures et les protéines déterminent la manière dont les signatures sont intégrées ensemble dans les entrées InterPro : le chevauchement comparatif des ensembles de protéines correspondantes et l’emplacement des correspondances des signatures sur les séquences sont utilisés comme indicateurs de parenté. Seules les signatures jugées d’une qualité suffisante sont intégrées dans InterPro. À partir de la version 81.0 (publiée le 21 août 2020), les entrées InterPro ont annoté 73,9% des résidus trouvés dans UniProtKB, avec 9,2% supplémentaires annotés par des signatures en attente d’intégration.

InterPro comprend également des données pour les variantes d’épissage et les protéines contenues dans les bases de données UniParc et UniMES.
Bases de données membres du consortium InterProEdit
Les signatures d’InterPro proviennent de 13 « bases de données membres », qui sont énumérées ci-dessous.
CATH-Gene3D Décrit les familles de protéines et les architectures de domaines dans les génomes complets. Les familles de protéines sont formées à l’aide d’un algorithme de clustering de Markov, suivi d’un clustering multi-linkage selon l’identité de séquence. Le mappage des domaines de structure et de séquence prédits est entrepris en utilisant des bibliothèques de modèles de Markov cachés représentant les domaines CATH et Pfam. Une annotation fonctionnelle est fournie aux protéines à partir de multiples ressources. La prédiction fonctionnelle et l’analyse des architectures de domaines sont disponibles sur le site Web de Gene3D. La CDD Conserved Domain Database est une ressource d’annotation des protéines qui consiste en une collection de modèles d’alignement de séquences multiples annotés pour des domaines anciens et des protéines complètes. Ces modèles sont disponibles sous forme de matrices de score spécifiques à la position (PSSM) pour une identification rapide des domaines conservés dans les séquences de protéines via RPS-BLAST. HAMAP signifie « High-quality Automated and Manual Annotation of microbial Proteomes » (annotation manuelle et automatisée de haute qualité des protéomes microbiens). Les profils HAMAP sont créés manuellement par des conservateurs experts qui identifient les protéines faisant partie de familles ou de sous-familles de protéines bactériennes, archéennes et codées par les plastides (c’est-à-dire les chloroplastes, les cyanelles, les apicoplastes, les plastides non photosynthétiques) bien conservées. MobiDB MobiDB est une base de données annotant le désordre intrinsèque des protéines. PANTHER PANTHER est une grande collection de familles de protéines qui ont été subdivisées en sous-familles fonctionnellement apparentées, en utilisant l’expertise humaine. Ces sous-familles modélisent la divergence des fonctions spécifiques au sein des familles de protéines, ce qui permet une association plus précise avec la fonction (classifications des fonctions moléculaires et des processus biologiques et diagrammes des voies de transmission établis par l’homme), ainsi que l’inférence des acides aminés importants pour la spécificité fonctionnelle. Des modèles de Markov cachés (HMM) sont construits pour chaque famille et sous-famille afin de classifier des séquences de protéines supplémentaires. Pfam Est une grande collection d’alignements de séquences multiples et de modèles de Markov cachés couvrant de nombreux domaines et familles de protéines communes.
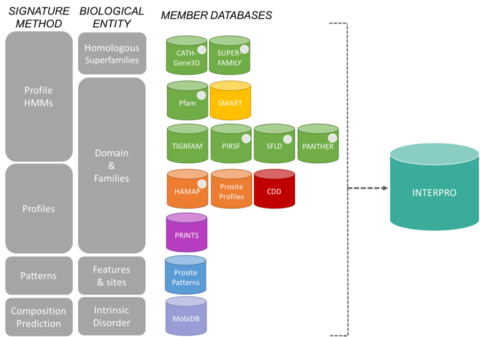
Le système de classification des protéines PIRSF est un réseau avec plusieurs niveaux de diversité de séquences, des superfamilles aux sous-familles, qui reflète la relation évolutive des protéines complètes et des domaines. La principale unité de classification PIRSF est la famille homéomorphe, dont les membres sont à la fois homologues (ayant évolué à partir d’un ancêtre commun) et homéomorphes (partageant une similarité de séquence pleine longueur et une architecture de domaine commune). PRINTS PRINTS est un recueil d’empreintes de protéines. Une empreinte est un groupe de motifs conservés utilisés pour caractériser une famille de protéines ; son pouvoir de diagnostic est affiné par un balayage itératif d’UniProt. En général, les motifs ne se chevauchent pas, mais sont séparés le long d’une séquence, bien qu’ils puissent être contigus dans l’espace 3D. Les empreintes digitales peuvent coder les plis et les fonctionnalités des protéines de manière plus souple et plus puissante que les motifs individuels, leur pleine puissance de diagnostic provenant du contexte mutuel fourni par les voisins des motifs. PROSITE PROSITE est une base de données de familles et de domaines de protéines. Elle est constituée de sites, de motifs et de profils biologiquement significatifs qui permettent d’identifier de manière fiable à quelle famille de protéines connue (le cas échéant) appartient une nouvelle séquence. SMART Simple Modular Architecture Research Tool Permet l’identification et l’annotation de domaines génétiquement mobiles et l’analyse des architectures de domaines. Plus de 800 familles de domaines trouvées dans les protéines de signalisation, extracellulaires et associées à la chromatine sont détectables. Ces domaines sont largement annotés en ce qui concerne les distributions phylétiques, la classe fonctionnelle, les structures tertiaires et les résidus fonctionnellement importants. SUPERFAMILY SUPERFAMILY est une bibliothèque de modèles de Markov cachés de profil qui représentent toutes les protéines de structure connue. La bibliothèque est basée sur la classification SCOP des protéines : chaque modèle correspond à un domaine SCOP et vise à représenter l’ensemble de la superfamille SCOP à laquelle le domaine appartient. SUPERFAMILY a été utilisé pour réaliser des affectations structurelles à tous les génomes complètement séquencés. SFLD Classification hiérarchique des enzymes qui associe des caractéristiques spécifiques de structure de séquence à des capacités chimiques spécifiques. TIGRFAMs TIGRFAMs est une collection de familles de protéines, comprenant des alignements de séquences multiples, des modèles de Markov cachés (HMM) et des annotations, qui fournit un outil pour identifier des protéines fonctionnellement apparentées sur la base de l’homologie de séquence. Les entrées qui sont des « équivalogues » regroupent des protéines homologues qui sont conservées en ce qui concerne la fonction.
Types de donnéesEdit
InterPro se compose de sept types de données fournies par différents membres du consortium :
| Type de données | Description | Bases de données contributrices | |
|---|---|---|---|
| Entrées InterPro | Structurelles. et/ou domaines fonctionnels de protéines prédits à l’aide d’une ou plusieurs signatures | Toutes les 13 bases de données membres | |
| Signatures des bases de données membres | Signatures des bases de données membres. Il s’agit notamment des signatures qui sont intégrées dans InterPro, et celles qui ne le sont pas | Les 13 bases de données membres | |
| Protéines | Séquences protéiques | UniProtKB (Swiss-Prot et TrEMBL) | |
| Protéome | Collection de protéines appartenant à un seul organisme | UniProtKB | |
| Structure | Structures tridimensionnelles des protéines | .dimensionnelles des protéines | PDBe |
| Taxonomie | Informations taxonomiques sur les protéines | UniProtKB | |
| Ensemble | Groupes de familles apparentées du point de vue de l’évolution | Pfam, CDD |

Types d’entrées InterProEdit
Les entrées InterPro peuvent être décomposées en cinq types :
- Superfamille homologue : Un groupe de protéines qui partagent une origine évolutive commune, comme en témoignent leurs similitudes structurelles, même si leurs séquences ne sont pas très similaires. Ces entrées ne sont spécifiquement fournies que par deux bases de données membres : CATH-Gene3D et SUPERFAMILY.
- Famille : Un groupe de protéines qui ont une origine évolutive commune déterminée par des similitudes structurelles, des fonctions connexes ou une homologie de séquence.
- Domaine : Une unité distincte dans une protéine avec une fonction, une structure ou une séquence particulière.
- Répétition : Une séquence d’acides aminés, généralement pas plus de 50 acides aminés, qui a tendance à se répéter de nombreuses fois dans une protéine.
- Site : Une courte séquence d’acides aminés où au moins un acide aminé est conservé. Il s’agit notamment des sites de modification post-traduction, des sites conservés, des sites de liaison et des sites actifs.