- Contenu
- Fonction
- Mécanisme d’inhibition de l’actio
- VIH et SIDA
- Impact de la structure
- Cristallisation de l’intasome du VFP
- Technique de cristallisation
- Statistiques cristallographiques et de raffinement
- Architecture globale & Composants
- Structure
- Intégrase et interactions avec l’ADN
- Site actif
- Inhibiteurs d’intégrase
Contenu
- 1 Fonction
- 2 Mécanisme d’actio…transfer inhibition actio
- 3 VIH et SIDA
- 4 Impact de la structure
- 5 Cristallisation de l’intasome du PFV
- 5.1 Technique de cristallisation
- 5.2 Statistiques cristallographiques et de raffinement
- 6 Architecture globale & Composants
- 6.1 Structure
- 6.2 Interactions entre l’intégrase et l’ADN
- 6.3 Site actif
- 7 Inhibiteurs d’intégrase
- 7.1 Ressources supplémentaires
- 8 Structures 3D de l’intégrase rétrovirale
Fonction
L’intégrase rétrovirale est une enzyme rétrovirale essentielle qui se lie à l’ADN viral et l’insère dans le chromosome d’une cellule hôte. L’ADNc transcrit en sens inverse du virus de l’immunodéficience humaine de type 1 (VIH-1) est inséré dans le génome de la cellule hôte afin d’augmenter la fitness et la virulence de l’agent pathogène. L’intégrase est produite par une classe de rétrovirus (comme le VIH) et est utilisée par le virus pour incorporer son matériel génétique dans l’ADN de la cellule hôte. La machinerie cellulaire de l’hôte produit alors l’ARNm puis les protéines à partir du matériel génétique incorporé, répliquant ainsi le virus. Bien que plusieurs médicaments inhibiteurs de l’intégrase aient été étudiés, le mécanisme responsable de l’action d’inhibition du transfert de brin reste à élucider. Cependant, Hare el al (2010) a déterminé les constituants structurels de l’intégration rétrovirale. Une élucidation plus poussée de la structure complète de l’intégrase rétrovirale, et son application à la régulation des activités fonctionnelles et enzymatiques pourraient potentiellement permettre aux chercheurs de retarder la progression des maladies rétrovirales. De plus, l’étude de l’intégration du VIH-1 pourrait conduire à une nouvelle cible prometteuse, et contribuer à la génération de modèles pharmacophores pour la thérapie antivirale.
Inhibiteurs d’intégrase du VIH : Le raltégravir, commercialisé sous le nom d’Isentress est actuellement approuvé comme inhibiteur thérapeutique de l’intégrase du VIH. Il a été approuvé le 12 octobre 2007.Pour l’intégrase phagique, voir Intégrase phagique.
Mécanisme d’inhibition de l’actio
Caption
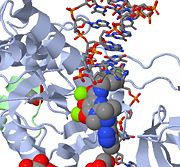
Supérieur est une image JMOL du MK-0518 bloquant l’extrémité 3′ de l’ADN viral pour l’empêcher de se lier au site actif. Remarquez les atomes d’oxygène chélateurs de métaux du MK-0518 interagissant avec les cations magnésium présents dans le site actif.
L’intégrase de type 1 du VIH est une cible relativement nouvelle et inédite pour les inhibiteurs. En 2007, le premier inhibiteur de l’intégrase du VIH-1, le Raltegravir, a été approuvé par la FDA pour une utilisation dans le VIH-1 en tant que thérapie combinée . Les inhibiteurs du transfert de brin agissent en empêchant l’intégration concertée de l’ADN viral dans le chromosome de l’hôte. Après l’entrée du virus dans la cellule hôte, la transcriptase inverse convertit l’ARNs viral en ADNdb. À ce stade, l’intégrase forme un complexe avec l’ADN viral, créant le complexe de pré-intégration (intasome). Le complexe de pré-intégration est ensuite chaperonné dans le noyau, où deux nucléotides sont excisés de l’extrémité 3′. Ensuite, l’ADN est intégré de manière covalente dans l’ADN hôte. Les inhibiteurs du transfert de brin interrompent ce processus, empêchant l’intégration de l’ADN viral dans le chromosome hôte. Les inhibiteurs de transfert de brin agissent en engageant les cofacteurs d’ions métalliques présents dans le site actif de l’intégrase rétrovirale. Les atomes d’oxygène chélateurs de métaux présents dans les inhibiteurs interagissent directement avec les cofacteurs métalliques, tandis que le groupe halobenzyle s’insère dans la poche créée par l’ADN viral 3′ déplacé dans le site actif.
VIH et SIDA

En 2010, il y a plus de 25 millions de personnes qui sont mortes du sida, et on estime qu’environ 33 millions de personnes vivent avec le VIH.
À ce jour, plus de 25 millions de personnes sont mortes du sida et on estime qu’environ 33 millions de personnes vivent avec le VIH aujourd’hui. Pour les inhibiteurs de l’intégrase rétrovirale, voir Raltegravir et Pharmacocinétique des inhibiteurs de l’intégrase rétrovirale.
Impact de la structure
Les structures tridimensionnelles de certaines protéines de la cellule hôte essentielles à la compréhension du mécanisme de l’infection par le VIH et de la virulence ont émergé des analyses cristallographiques aux rayons X. Les structures de la protéase et de l’intégrase du VIH figurent parmi les structures les mieux classées qui ont contribué à sauver de nombreuses vies et à améliorer la qualité de vie de nombreuses personnes atteintes du VIH. Elle est mise en œuvre dans la conception de médicaments basée sur la structure pour développer des inhibiteurs de protéase et des inhibiteurs d’intégrase, et est utilisée comme un composant important de la thérapie antirétrovirale hautement active (HAART).
Bien que les agents antirétroviraux existants améliorent la qualité de vie et prolongent la vie de nombreux patients, ils ne parviennent pas à éradiquer la maladie. Des études sur les inhibiteurs d’intégrase montrent que la combinaison avec d’autres médicaments antirétroviraux diminue les adaptations virales, et pourrait avoir le potentiel d’être utilisé pour une thérapie de sauvetage pour les patients qui ont acquis une résistance aux autres médicaments. Pour en savoir plus, veuillez consulter
- Le sida avant les inhibiteurs de protéase &Inhibiteurs de protéase du VIH : Une percée
- Traitements:Inhibiteur d’intégrase rétroviral Pharmacocinétique Références.
Cristallisation de l’intasome du VFP
Pour imiter les extrémités de l’ADN viral du VIH-1, Hare et al (2010) ont utilisé des préparations solubles et entièrement fonctionnelles de l’intasome du prototype du virus spumeux (VFP), obtenues à l’aide d’intégrase recombinante du VFP et d’oligonucléotides à double brin.
La remarquable stabilité des complexes intégrase-ADN a été déterminée en observant les réactions de transfert de brin in vitro, qui ont été classées en trois modes de migration de déprotection : (1) événements concertés uniques : plasmide cible linéarisé ; (2) événements concertés multiples : frottis ; (3) événements en demi-site : ADN circulaire ouvert. Une caractérisation plus poussée de l’intasome du VFP a également montré une substantialité structurelle qui implique de fortes interactions protéine-protéine et protéine-ADN malgré une incubation prolongée dans des conditions de force ionique élevée. Des essais de cristallisation complets ont permis d’obtenir une configuration cristalline viable qui a diffracté les rayons X avec une résolution de 2,9 angströms. Une structure tridimensionnelle a finalement été déterminée. L’unité asymétrique contient un seul dimère d’intégrase avec une molécule d’ADN viral liée de façon stable, et une paire de dimères d’intégrase associés à la symétrie, qui forment un tétramère oblong. L’interface du dimère est stabilisée par des interactions intermoléculaires entre les domaines amino terminaux et le noyau catalytique (sous-unité interne – sous-unité externe). La forme globale du tétramère oblong est unique bien que présentant des semblants de complexes d’intégrase du VIH-1 précédemment rapportés.
Technique de cristallisation
Les complexes protéine-ADN ont été formés en utilisant l’IN du VFP de type sauvage pleine longueur et un ADNdb synthétique qui modélisait l’extrémité virale.
L’intasome a été cristallisé en utilisant la technique de diffusion suspendue à goutte de vapeur. La solution réservoir était composée de sulfate d’ammonium 1,35 M, de glycérol à 25 % (v/v), de 1,6-hexanediol à 4,8 % (v/v) et d’acide 2-(N-morpholino) éthanesulfonique (MES) 50 mM à pH 6,5. Les cristaux de protéine-ADN ont également été trempés en présence de MK0518, GS9137, Mg(II), et/ou Mn(II). La structure cristalline a été résolue en utilisant le remplacement moléculaire.
Statistiques cristallographiques et de raffinement
Hare et al (2010) ont publié des données sur sept structures cristallines. Ces données comprennent le complexe PFV IN (forme apo) et six structures supplémentaires, y compris le complexe lié à Mg, Mn, Mg/MK0518, Mn/MK0518, Mg/GS9137 et Mn/GS9137. Les sept structures appartiennent au groupe spatial P41212. Elles ont été affinées à une résolution comprise entre 2,85 et 3,25 Å.
Architecture globale & Composants
Structure
La structure globale de l’intasome VFP assemblé est un modèle de tétramère basé sur deux structures de domaine avec une interface dimère-dimère. Les modèles d’intasome précédents dépeignent une structure similaire mais plus flexible alors que l’intasome du VFP s’est avéré être fortement contraint. En utilisant la modélisation de l’homologie, Hare et al (2010) proposent que des lieurs interdomaines plus courts puissent être un facteur de flexibilité, spécifiquement dans l’intégrase du VIH-1. Les sous-unités internes du tétramère sont impliquées dans la tétramérisation globale et la liaison de l’ADN viral. Les domaines du noyau catalytique des sous-unités externes peuvent agir comme des supports, mais comme les domaines amino- et carboxy-terminaux ne sont pas résolus dans les cartes de densité électronique, leur fonction reste peu concluante. Le domaine central catalytique et le lieur du domaine carboxy-terminal adoptent une conformation étendue sur la majeure partie de leur longueur, et sont situés parallèlement au domaine amino-terminal et au lieur du domaine central catalytique de la sous-unité interne. Les lieurs interdomaines Les lieurs interdomaines (lieur CCD-CTD et lieur NTD-CCD) lient les deux moitiés de l’intasome ensemble, et la structure est encore stabilisée par une paire de domaines carboxy-terminaux interagissant avec les deux domaines de noyau catalytique internes.
Intégrase et interactions avec l’ADN
De fortes interactions protéine-ADN sont situées dans les six nucléotides terminaux. Chaque domaine carboxy-terminal interagit avec le squelette phosphodiester des deux molécules d’ADN viral. De plus, le domaine d’extension du domaine amino-terminal et le domaine amino-terminal interagissent avec l’ADN viral au niveau du site actif du domaine de noyau catalytique opposé.
Site actif
Les carboxylates du site actif sont des chaînes latérales d’Asp 128, Asp 185, Glu 221.
Un atome de zinc chacun est situé près des sites actifs.
- .
- .
Inhibiteurs d’intégrase
| Nom | Marque | Société | Brevet | Notes |
| Raltegravir | Isentress | Merck & Co. | – | également connu sous le nom de MK-0518. L’isopropyl et le méthyl-oxadiazole du MK-0518 sont impliqués dans des interactions hydrophobes et d’empilement avec les chaînes latérales de Pro 214 et Tyr 212 pour stabiliser ce médicament au sein du site actif de l’intasome du VFP. Cette façon de se lier au médicament entraîne le déplacement de l’extrémité 3′ réactive de l’ADN viral du site actif de l’intasome du VFP. Après la liaison du MK-0518 au site actif, le groupe hydroxyle 3′ réactif s’éloigne du site actif de l’intasome du VFP de plus de 6 Angstroms. Le raltégravir a été approuvé par la FDA le 12 octobre 2007 pour être utilisé avec d’autres agents anti-VIH dans le traitement de l’infection par le VIH chez les adultes. C’est le premier inhibiteur d’intégrase approuvé par la FDA. |
| Elvitegravir | – | Gilead Science | – | GS-9137 interagit avec le Pro 214 de l’intasome du VFP par l’intermédiaire de sa base quinolone et de son groupe isopropyle. Aux stades expérimentaux ; partage la structure de base des antibiotiques quinolones. Les études de phase II de l’elvitegravir chez les personnes ayant déjà été traitées sont terminées. Des études de phase III chez des patients ayant déjà reçu un traitement sont en cours. Une étude de phase II de l’elvitégravir chez des personnes n’ayant jamais suivi de traitement antirétroviral est en cours. Cette étude évaluera également un agent de renforcement à la place du Norvir, actuellement appelé GS9350. L’elvitegravir est prometteur pour les patients séropositifs qui ont déjà pris d’autres médicaments anti-VIH. |
| MK-2048 | – | Merck & Co. | – | Inhibiteur d’intégrase de deuxième génération, destiné à être utilisé contre l’infection par le VIH. Il est supérieur au premier inhibiteur d’intégrase disponible, le raltégravir, car il inhibe l’enzyme VIH intégrase 4 fois plus longtemps. Il est à l’étude pour être utilisé dans le cadre d’une prophylaxie pré-exposition (PrEP). |
Voir aussi Rétrovirus Inhibiteur d’intégrase Pharmacocinétique.