InterPro enthält drei Haupteinheiten: Proteine, Signaturen (auch als „Methoden“ oder „Modelle“ bezeichnet) und Einträge. Die Proteine in UniProtKB sind auch die zentralen Proteineinheiten in InterPro. Informationen darüber, welche Signaturen signifikant mit diesen Proteinen übereinstimmen, werden berechnet, wenn die Sequenzen von UniProtKB freigegeben werden, und diese Ergebnisse werden der Öffentlichkeit zur Verfügung gestellt (siehe unten). Die Übereinstimmungen der Signaturen mit den Proteinen sind ausschlaggebend dafür, wie die Signaturen in die InterPro-Einträge integriert werden: Die vergleichende Überlappung der übereinstimmenden Proteinsätze und die Position der Übereinstimmungen der Signaturen auf den Sequenzen werden als Indikatoren für die Verwandtschaft verwendet. Nur Signaturen, deren Qualität als ausreichend erachtet wird, werden in InterPro integriert. Zum Zeitpunkt der Version 81.0 (freigegeben am 21. August 2020) wurden 73,9 % der in UniProtKB gefundenen Reste durch InterPro-Einträge annotiert, weitere 9,2 % wurden durch Signaturen annotiert, deren Integration noch aussteht.

InterPro enthält auch Daten für Spleißvarianten und die in den Datenbanken UniParc und UniMES enthaltenen Proteine.
Mitgliedsdatenbanken des InterPro-KonsortiumsBearbeiten
Die Signaturen von InterPro stammen aus 13 „Mitgliedsdatenbanken“, die nachstehend aufgeführt sind.
CATH-Gene3D Beschreibt Proteinfamilien und Domänenarchitekturen in vollständigen Genomen. Proteinfamilien werden mit Hilfe eines Markov-Clustering-Algorithmus gebildet, gefolgt von Multi-Linkage-Clustering nach Sequenzidentität. Das Mapping der vorhergesagten Struktur- und Sequenzdomänen erfolgt mithilfe von Hidden-Markov-Modell-Bibliotheken, die CATH- und Pfam-Domänen darstellen. Funktionelle Annotationen werden für Proteine aus verschiedenen Quellen bereitgestellt. Funktionelle Vorhersagen und Analysen von Domänenarchitekturen sind auf der Gene3D-Website verfügbar. CDD Conserved Domain Database ist eine Ressource für die Annotation von Proteinen, die aus einer Sammlung von annotierten Multiple-Sequence-Alignment-Modellen für alte Domänen und Proteine in voller Länge besteht. Diese sind als positionsspezifische Score-Matrizen (PSSMs) für die schnelle Identifizierung von konservierten Domänen in Proteinsequenzen über RPS-BLAST verfügbar. HAMAP steht für High-quality Automated and Manual Annotation of microbial Proteomes. HAMAP-Profile werden von Experten manuell erstellt und identifizieren Proteine, die zu gut konservierten bakteriellen, archaeischen und plastidenkodierten (d. h. Chloroplasten, Cyanellen, Apicoplasten, nichtphotosynthetische Plastiden) Proteinfamilien oder -unterfamilien gehören. MobiDB MobiDB ist eine Datenbank, die intrinsische Unordnung in Proteinen annotiert. PANTHER PANTHER ist eine große Sammlung von Proteinfamilien, die mit Hilfe menschlichen Fachwissens in funktionell verwandte Unterfamilien unterteilt wurden. Diese Unterfamilien modellieren die Divergenz spezifischer Funktionen innerhalb von Proteinfamilien und ermöglichen eine genauere Zuordnung zu Funktionen (von Menschen kuratierte Klassifizierungen von molekularen Funktionen und biologischen Prozessen sowie Pfaddiagramme) sowie Rückschlüsse auf Aminosäuren, die für die funktionelle Spezifität wichtig sind. Hidden-Markov-Modelle (HMMs) werden für jede Familie und Unterfamilie erstellt, um zusätzliche Proteinsequenzen zu klassifizieren. Pfam ist eine große Sammlung von Mehrfachsequenz-Alignments und Hidden-Markov-Modellen, die viele gängige Proteindomänen und -familien abdecken.
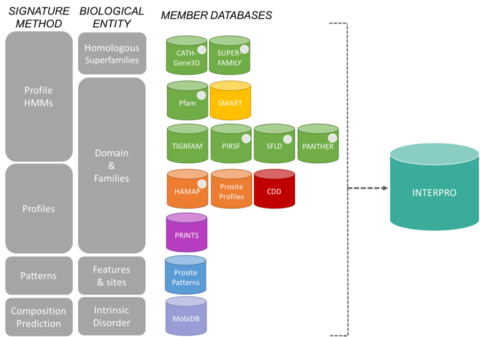
Das PIRSF-Proteinklassifizierungssystem ist ein Netzwerk mit mehreren Ebenen der Sequenzvielfalt von Superfamilien bis zu Unterfamilien, das die evolutionäre Beziehung von Proteinen in voller Länge und Domänen widerspiegelt. Die primäre PIRSF-Klassifizierungseinheit ist die homomorphe Familie, deren Mitglieder sowohl homolog (von einem gemeinsamen Vorfahren abstammend) als auch homomorph (mit ähnlicher Sequenz in voller Länge und einer gemeinsamen Domänenarchitektur) sind. PRINTS PRINTS ist ein Kompendium von Protein-Fingerabdrücken. Ein Fingerabdruck ist eine Gruppe von konservierten Motiven, die zur Charakterisierung einer Proteinfamilie verwendet werden; seine diagnostische Leistung wird durch iteratives Scannen von UniProt verfeinert. In der Regel überschneiden sich die Motive nicht, sondern sind entlang einer Sequenz getrennt, obwohl sie im 3D-Raum zusammenhängend sein können. Fingerabdrücke können Proteinfalten und -funktionalitäten flexibler und leistungsfähiger kodieren als einzelne Motive, wobei sich ihre volle diagnostische Kraft aus dem gegenseitigen Kontext ergibt, den die Motivnachbarn bieten. PROSITE PROSITE ist eine Datenbank von Proteinfamilien und -domänen. Sie besteht aus biologisch bedeutsamen Stellen, Mustern und Profilen, mit deren Hilfe sich zuverlässig feststellen lässt, zu welcher bekannten Proteinfamilie (falls vorhanden) eine neue Sequenz gehört. SMART Simple Modular Architecture Research Tool Ermöglicht die Identifizierung und Annotation von genetisch mobilen Domänen und die Analyse von Domänenarchitekturen. Mehr als 800 Domänenfamilien, die in Signalproteinen, extrazellulären und Chromatin-assoziierten Proteinen vorkommen, sind nachweisbar. Diese Domänen sind hinsichtlich der phyletischen Verteilung, der funktionellen Klasse, der Tertiärstrukturen und der funktionell wichtigen Reste ausführlich annotiert. SUPERFAMILY SUPERFAMILY ist eine Bibliothek von versteckten Markov-Profilmodellen, die alle Proteine mit bekannter Struktur repräsentieren. Die Bibliothek basiert auf der SCOP-Klassifikation von Proteinen: Jedes Modell entspricht einer SCOP-Domäne und soll die gesamte SCOP-Superfamilie repräsentieren, zu der die Domäne gehört. SUPERFAMILY wurde verwendet, um strukturelle Zuordnungen zu allen vollständig sequenzierten Genomen vorzunehmen. SFLD Eine hierarchische Klassifizierung von Enzymen, die bestimmte Sequenzstrukturmerkmale mit bestimmten chemischen Fähigkeiten in Beziehung setzt. TIGRFAMs TIGRFAMs ist eine Sammlung von Proteinfamilien mit kuratierten multiplen Sequenzalignments, versteckten Markov-Modellen (HMMs) und Annotation, die ein Werkzeug zur Identifizierung funktionell verwandter Proteine auf der Grundlage von Sequenzhomologie darstellt. Die Einträge, die „equivalogs“ sind, gruppieren homologe Proteine, die in Bezug auf die Funktion konserviert sind.
DatentypenBearbeiten
InterPro besteht aus sieben Arten von Daten, die von verschiedenen Mitgliedern des Konsortiums bereitgestellt werden:
| Datentyp | Beschreibung | Beitragende Datenbanken |
|---|---|---|
| InterPro-Einträge | Strukturelle und/oder funktionelle Domänen von Proteinen, die mit Hilfe einer oder mehrerer Signaturen vorhergesagt wurden | Alle 13 Mitgliedsdatenbanken |
| Signaturen von Mitgliedsdatenbanken | Signaturen von Mitgliedsdatenbanken. Dazu gehören Signaturen, die in InterPro integriert sind, und solche, die nicht integriert sind | Alle 13 Mitgliedsdatenbanken |
| Protein | Proteinsequenzen | UniProtKB (Swiss-Prot und TrEMBL) |
| Proteom | Sammlung von Proteinen, die zu einem einzigen Organismus gehören | UniProtKB |
| Struktur | 3-dimensionalen Strukturen von Proteinen | PDBe |
| Taxonomie | Taxonomische Informationen über Proteine | UniProtKB |
| Set | Gruppen von evolutionär verwandten Familien | Pfam, CDD |

InterPro-EintragstypenBearbeiten
InterPro-Einträge können weiter in fünf Typen unterteilt werden:
- Homologe Superfamilie: Eine Gruppe von Proteinen, die einen gemeinsamen evolutionären Ursprung haben, der sich in ihren strukturellen Ähnlichkeiten zeigt, auch wenn ihre Sequenzen nicht sehr ähnlich sind. Diese Einträge werden speziell nur von zwei Mitgliedsdatenbanken bereitgestellt: CATH-Gene3D und SUPERFAMILY.
- Familie: Eine Gruppe von Proteinen, die einen gemeinsamen evolutionären Ursprung haben, der durch strukturelle Ähnlichkeiten, verwandte Funktionen oder Sequenzhomologie bestimmt wird.
- Domäne: Eine ausgeprägte Einheit in einem Protein mit einer bestimmten Funktion, Struktur oder Sequenz.
- Wiederholung: Eine Sequenz von Aminosäuren, in der Regel nicht länger als 50 Aminosäuren, die dazu neigt, sich viele Male in einem Protein zu wiederholen.
- Stelle: Eine kurze Sequenz von Aminosäuren, in der mindestens eine Aminosäure konserviert ist. Dazu gehören posttranslationale Modifikationsstellen, konservierte Stellen, Bindungsstellen und aktive Stellen.