InterPro contiene tres entidades principales: proteínas, firmas (también denominadas «métodos» o «modelos») y entradas. Las proteínas de UniProtKB son también las entidades proteicas centrales de InterPro. La información relativa a las firmas que coinciden significativamente con estas proteínas se calcula a medida que las secuencias son liberadas por UniProtKB y estos resultados se ponen a disposición del público (véase más adelante). Las coincidencias de las firmas con las proteínas son las que determinan cómo se integran las firmas en las entradas de InterPro: el solapamiento comparativo de los conjuntos de proteínas coincidentes y la ubicación de las coincidencias de las firmas en las secuencias se utilizan como indicadores de parentesco. Sólo se integran en InterPro las firmas que se consideran de suficiente calidad. A partir de la versión 81.0 (publicada el 21 de agosto de 2020) las entradas de InterPro anotaron el 73,9% de los residuos encontrados en UniProtKB con otro 9,2% anotado por firmas que están pendientes de integración.

InterPro también incluye datos de variantes de empalme y de las proteínas contenidas en las bases de datos UniParc y UniMES.
Bases de datos de miembros del consorcio InterProEditar
Las firmas de InterPro provienen de 13 «bases de datos de miembros», que se enumeran a continuación.
CATH-Gene3D Describe familias de proteínas y arquitecturas de dominios en genomas completos. Las familias de proteínas se forman mediante un algoritmo de agrupación de Markov, seguido de una agrupación de enlaces múltiples según la identidad de la secuencia. El mapeo de la estructura predicha y los dominios de la secuencia se lleva a cabo utilizando bibliotecas de modelos de Markov ocultos que representan los dominios CATH y Pfam. Se proporciona una anotación funcional a las proteínas a partir de múltiples recursos. La predicción funcional y el análisis de las arquitecturas de los dominios están disponibles en el sitio web Gene3D. La base de datos de dominios conservados CDD es un recurso de anotación de proteínas que consiste en una colección de modelos de alineación de secuencias múltiples anotados para dominios antiguos y proteínas de longitud completa. Están disponibles como matrices de puntuación de posición específica (PSSM) para la identificación rápida de dominios conservados en secuencias de proteínas mediante RPS-BLAST. HAMAP son las siglas de High-quality Automated and Manual Annotation of microbial Proteomes. Los perfiles HAMAP son creados manualmente por curadores expertos que identifican proteínas que forman parte de familias o subfamilias de proteínas bien conservadas de bacterias, arqueas y plastos (es decir, cloroplastos, cianelas, apicoplastos, plastos no fotosintéticos). MobiDB MobiDB es una base de datos que anota el desorden intrínseco de las proteínas. PANTHER PANTHER es una gran colección de familias de proteínas que han sido subdivididas en subfamilias funcionalmente relacionadas, utilizando la experiencia humana. Estas subfamilias modelan la divergencia de funciones específicas dentro de las familias de proteínas, lo que permite una asociación más precisa con la función (clasificaciones de funciones moleculares y procesos biológicos curadas por humanos y diagramas de vías), así como la inferencia de aminoácidos importantes para la especificidad funcional. Los modelos ocultos de Markov (HMM) se construyen para cada familia y subfamilia para clasificar las secuencias de proteínas adicionales. Pfam Es una gran colección de alineaciones de secuencias múltiples y modelos de Markov ocultos que cubren muchos dominios y familias de proteínas comunes.
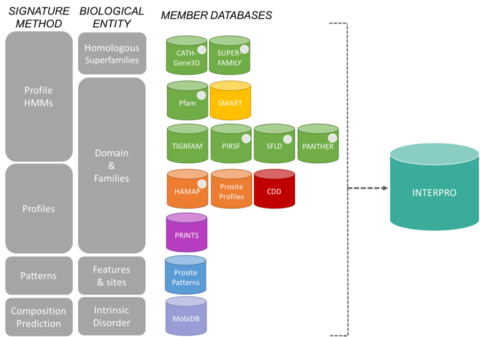
El sistema de clasificación de proteínas PIRSF es una red con múltiples niveles de diversidad de secuencias, desde superfamilias a subfamilias, que refleja la relación evolutiva de las proteínas de longitud completa y los dominios. La principal unidad de clasificación de PIRSF es la familia homeomórfica, cuyos miembros son a la vez homólogos (evolucionaron a partir de un ancestro común) y homeomórficos (comparten la similitud de la secuencia de longitud completa y una arquitectura de dominio común). PRINTS PRINTS es un compendio de huellas digitales de proteínas. Una huella dactilar es un grupo de motivos conservados que se utilizan para caracterizar una familia de proteínas; su poder de diagnóstico se perfecciona mediante el escaneo iterativo de UniProt. Por lo general, los motivos no se solapan, sino que están separados a lo largo de una secuencia, aunque pueden ser contiguos en el espacio 3D. Las huellas dactilares pueden codificar los pliegues y las funcionalidades de las proteínas de forma más flexible y potente que los motivos individuales, ya que toda su potencia de diagnóstico se deriva del contexto mutuo que ofrecen los motivos vecinos. PROSITE PROSITE es una base de datos de familias y dominios de proteínas. Consta de sitios, patrones y perfiles biológicamente significativos que ayudan a identificar de forma fiable a qué familia de proteínas conocida (si la hay) pertenece una nueva secuencia. SMART Simple Modular Architecture Research Tool Permite la identificación y anotación de dominios genéticamente móviles y el análisis de arquitecturas de dominios. Se pueden detectar más de 800 familias de dominios encontrados en proteínas de señalización, extracelulares y asociadas a la cromatina. Estos dominios están ampliamente anotados con respecto a las distribuciones filéticas, la clase funcional, las estructuras terciarias y los residuos funcionalmente importantes. SUPERFAMILIA SUPERFAMILIA es una biblioteca de modelos de Markov ocultos de perfil que representan todas las proteínas de estructura conocida. La biblioteca se basa en la clasificación SCOP de las proteínas: cada modelo corresponde a un dominio SCOP y pretende representar toda la superfamilia SCOP a la que pertenece el dominio. SUPERFAMILY se ha utilizado para realizar asignaciones estructurales a todos los genomas completamente secuenciados. SFLD Clasificación jerárquica de las enzimas que relaciona características específicas de la estructura de la secuencia con capacidades químicas específicas. TIGRFAMs TIGRFAMs es una colección de familias de proteínas, con alineaciones de secuencias múltiples curadas, modelos de Markov ocultos (HMM) y anotaciones, que proporciona una herramienta para identificar proteínas funcionalmente relacionadas en base a la homología de la secuencia. Las entradas que son «equivalentes» agrupan proteínas homólogas que se conservan con respecto a la función.
Tipos de datosEditar
InterPro consta de siete tipos de datos proporcionados por diferentes miembros del consorcio:
| Tipo de datos | Descripción | Bases de datos contribuyentes |
|---|---|---|
| Entradas de InterPro | Dominios estructurales y/o dominios funcionales de proteínas predichas utilizando una o más firmas | Todas las 13 bases de datos miembros |
| Firmas de bases de datos miembros | Firmas de bases de datos miembros. Estas incluyen las firmas que están integradas en InterPro, y las que no lo están | Todas las 13 bases de datos miembros |
| Secuencias de proteínas | UniProtKB (Swiss-Prot y TrEMBL) | |
| Proteoma | Colección de proteínas que pertenecen a un solo organismo | UniProtKB |
| Estructura | 3-estructuras dimensionales de las proteínas | PDBe |
| Taxonomía | Información taxonómica de las proteínas | UniProtKB |
| Set | Grupos de familias relacionadas evolutivamente | Pfam, CDD |

Tipos de entradas de InterProEditar
Las entradas de InterPro pueden desglosarse en cinco tipos:
- Superfamilia homóloga: Un grupo de proteínas que comparten un origen evolutivo común como se ve en sus similitudes estructurales, incluso si sus secuencias no son altamente similares. Estas entradas sólo son proporcionadas específicamente por dos bases de datos miembros: CATH-Gene3D y SUPERFAMILY.
- Familia: Un grupo de proteínas que tienen un origen evolutivo común determinado a través de similitudes estructurales, funciones relacionadas u homología de secuencia.
- Dominio: Una unidad distinta en una proteína con una función, estructura o secuencia particular.
- Repetición: Una secuencia de aminoácidos, normalmente de no más de 50 aminoácidos, que tienden a repetirse muchas veces en una proteína.
- Sitio: Una secuencia corta de aminoácidos en la que se conserva al menos un aminoácido. Entre ellos se encuentran los sitios de modificación post-traducción, los sitios conservados, los sitios de unión y los sitios activos.