- Contenidos
- Función
- Mecanismo de la acción de inhibición de la transferencia de cadena
- VIH y sida
- Impacto de la estructura
- Cristalización del intasoma del PFV
- Técnica de cristalización
- Estadísticas cristalográficas y de refinamiento
- Arquitectura general & Componentes
- Estructura
- Interacciones entre la integrasa y el ADN
- Sitio activo
- Inhibidores de la integrasa
Contenidos
- 1 Función
- 2 Mecanismo de inhibición de la cadena detransferencia de actio
- 3 VIH y SIDA
- 4 Impacto de la estructura
- 5 Cristalización del intasoma del PFV
- 5.1 Técnica de cristalización
- 5.2 Estadísticas cristalográficas y de refinamiento
- 6 Arquitectura global &Componentes
- 6.1 Estructura
- 6.2 Interacciones entre la integrasa y el ADN
- 6.3 Sitio activo
- 7 Inhibidores de la integrasa
- 7.1 Recursos adicionales
- 8 Estructuras 3D de la integrasa retroviral
Función
La integrasa retroviral es una enzima retroviral esencial que se une al ADN viral y lo inserta en un cromosoma de la célula huésped. El ADNc transcrito inversamente del virus de la inmunodeficiencia humana tipo 1 (VIH-1) se inserta en el genoma de la célula huésped para aumentar la aptitud y virulencia del patógeno. La integrasa es producida por una clase de retrovirus (como el VIH) y es utilizada por el virus para incorporar su material genético al ADN de la célula huésped. La maquinaria celular del huésped produce entonces ARNm y luego proteínas a partir del material genético incorporado, replicando así el virus. Aunque se han investigado varios fármacos inhibidores de la integrasa, aún no se ha dilucidado el mecanismo responsable de la acción inhibidora de la transferencia de cadena. Sin embargo, Hare el al (2010) determinó los componentes estructurales de la integración retroviral. Una mayor elucidación de la estructura completa de la integrasa retroviral y su aplicación para regular las actividades funcionales y enzimáticas podría permitir a los investigadores retrasar la progresión de las enfermedades retrovirales. Además, el estudio de la integración del VIH-1 podría conducir a una nueva y prometedora diana, y contribuir a la generación de modelos farmacológicos para la terapia antiviral.
Inhibidores de la integrasa del VIH: Raltegravir, comercializado como Isentress, está actualmente aprobado como inhibidor terapéutico de la integrasa del VIH. Fue aprobado el 12 de octubre de 2007.Para la integrasa fágica véase Integrasa fágica.
Mecanismo de la acción de inhibición de la transferencia de cadena
Caption
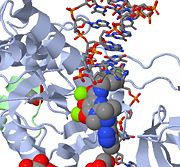
Arriba se muestra una imagen JMOL del MK-0518 bloqueando el extremo 3′ del ADN viral para que no se una al sitio activo. Obsérvese que los átomos de oxígeno quelantes del MK-0518 interactúan con los cationes de magnesio que se encuentran en el sitio activo.
La integrasa del VIH tipo 1 es un objetivo relativamente nuevo y novedoso para los inhibidores. En 2007, el primer inhibidor de la integrasa del VIH-1, Raltegravir, fue aprobado por la FDA para su uso en el VIH-1 como terapia combinada . Los inhibidores de la transferencia de cadena actúan impidiendo la integración concertada del ADN viral en el cromosoma del huésped. Tras la entrada del virus en la célula huésped, la transcriptasa inversa convierte el ssRNA viral en dsDNA. En este punto, la integrasa forma un complejo con el ADN viral, creando el complejo de pre-integración (intasoma). A continuación, el complejo de preintegración es conducido al núcleo, donde se eliminan dos nucleótidos del extremo 3′. A continuación, el ADN se integra covalentemente en el ADN huésped. Los inhibidores de la transferencia de cadena interrumpen este proceso, impidiendo la integración del ADN viral en el cromosoma del huésped. Los inhibidores de la transferencia de la cadena actúan enganchando los cofactores de iones metálicos que se encuentran en el sitio activo de la integrasa retroviral. Los átomos de oxígeno quelantes del metal que se encuentran en los inhibidores interactúan directamente con los cofactores metálicos, mientras que el grupo halobencil encaja en el bolsillo creado por el ADN viral 3′ desplazado en el sitio activo.
VIH y sida

En 2010, hay más de 25 millones de personas que han muerto de SIDA, y se estima que aproximadamente 33 millones de personas viven con el VIH.
Hasta la fecha, más de 25 millones de personas han muerto de SIDA y se estima que aproximadamente 33 millones de personas viven con el VIH en la actualidad. Para los inhibidores de la integrasa retroviral, véase Raltegravir y Farmacocinética de los inhibidores de la integrasa retroviral.
Impacto de la estructura
A partir de los análisis cristalográficos de rayos X han surgido estructuras tridimensionales para ciertas proteínas de la célula huésped fundamentales para comprender el mecanismo de la infección y la virulencia del VIH. Las estructuras de la proteasa y la integrasa del VIH se encuentran entre las más valoradas y han contribuido a salvar muchas vidas y a mejorar la calidad de vida de muchas personas afectadas por el VIH. Se aplica en el diseño de fármacos basado en la estructura para desarrollar inhibidores de la proteasa e inhibidores de la integrasa, y se utiliza como un componente importante de la terapia antirretroviral altamente activa (HAART).
Aunque los agentes antirretrovirales existentes mejoran la calidad de vida y prolongan la vida de muchos pacientes, no consiguen erradicar la enfermedad. Los estudios sobre los inhibidores de la integrasa demuestran que la combinación con otros fármacos antirretrovirales disminuye las adaptaciones virales, y puede tener el potencial de utilizarse para la terapia de rescate de los pacientes que han adquirido resistencia a otros fármacos. Para más información, véase
- El sida antes de los inhibidores de la proteasa & Inhibidores de la proteasa del VIH: Un gran avance
- Tratamientos:Farmacocinética de los inhibidores de la integrasa retroviral Referencias.
Cristalización del intasoma del PFV
Para imitar los extremos del ADN viral del VIH-1, Hare et al (2010) utilizaron preparaciones del intasoma del virus espumoso prototipo (PFV) solubles y totalmente funcionales, obtenidas mediante la integrasa recombinante del PFV y oligonucleótidos de doble cadena.
La notable estabilidad de los complejos integrasa-ADN se determinó mediante la observación de las reacciones de transferencia de hebra in vitro, que se clasificaron en tres modos de migración de desproteinización (1) eventos concertados únicos: plásmido diana linealizado; (2) eventos concertados múltiples: mancha; (3) eventos de medio sitio: ADN circular abierto. La caracterización adicional del intasoma del VFP también mostró una sustancialidad estructural que implicaba fuertes interacciones proteína-proteína y proteína-ADN a pesar de la incubación prolongada en condiciones de alta fuerza iónica. Los ensayos de cristalización exhaustivos permitieron obtener una configuración cristalina viable que difractaba los rayos X con una resolución de 2,9 Angstroms. Finalmente se determinó una estructura tridimensional. La unidad asimétrica contenía un único dímero de integrasa con una molécula de ADN viral unida de forma estable, y un par de dímeros de integrasa consociados con simetría, que formaban un tetrámero oblongo. La interfaz del dímero está estabilizada por las interacciones intermoleculares de los dominios aminoterminales y del núcleo catalítico (subunidad interna-subunidad externa). La forma general del tetrámero oblongo es única, aunque guarda similitudes con los complejos de integrasa del VIH-1 descritos anteriormente.
Técnica de cristalización
Los complejos proteína-ADN se formaron utilizando el PFV IN de longitud completa, de tipo salvaje, y ADNd sintético que modelaba el extremo viral.
El intasoma se cristalizó utilizando la técnica de difusión colgante por goteo de vapor. La solución de reserva consistía en 1,35 M de sulfato de amonio, 25% (v/v) de glicerol, 4,8% (v/v) de 1,6-hexanediol y 50mM de ácido 2-(N-morfolino) etanosulfónico (MES) a pH 6,5. Los cristales de proteína-ADN también se empaparon en presencia de MK0518, GS9137, Mg(II), y/o Mn(II). La estructura cristalina se resolvió utilizando el reemplazo molecular.
Estadísticas cristalográficas y de refinamiento
Hare et al (2010) han publicado datos sobre siete estructuras cristalinas. Estos datos incluyen el complejo PFV IN (forma apo) y seis estructuras adicionales, incluyendo el complejo unido a Mg, Mn, Mg/MK0518, Mn/MK0518, Mg/GS9137 y Mn/GS9137. Las siete estructuras pertenecen al grupo espacial P41212. Se han refinado a una resolución de entre 2,85 y 3,25 Å.
Arquitectura general & Componentes
Estructura
La estructura general del intasoma PFV ensamblado es un modelo de tetrámero basado en dos estructuras de dominio con una interfaz de dímero. Los modelos anteriores del intasoma describen una estructura similar pero más flexible, mientras que el intasoma del PFV ha demostrado estar muy restringido. Utilizando modelos de homología, Hare et al (2010) proponen que los enlazadores interdominio más cortos pueden ser un factor de flexibilidad, específicamente en la integrasa del VIH-1. Las subunidades internas del tetrámero están implicadas en la tetramerización general y en la unión del ADN viral. Los dominios del núcleo catalítico de las subunidades externas pueden actuar como soportes, pero como los dominios amino y carboxi-terminal no están resueltos en los mapas de densidad electrónica, su función sigue sin ser concluyente. El dominio central catalítico y el enlazador del dominio carboxi-terminal adoptan una conformación extendida en la mayor parte de su longitud, y se sitúan en paralelo al dominio amino-terminal y al enlazador del dominio central catalítico de la subunidad interna. Los enlazadores interdominio Los enlazadores interdominio (enlazador CCD-CTD y enlazador NTD-CCD) unen ambas mitades del intasoma, y la estructura se estabiliza aún más por un par de dominios carboxi-terminales que interactúan con ambos dominios del núcleo catalítico interno.
Interacciones entre la integrasa y el ADN
Las fuertes interacciones proteína-ADN se localizan en los seis nucleótidos terminales. Cada dominio carboxi-terminal interactúa con el esqueleto fosfodiéster de ambas moléculas de ADN viral. Además, el dominio de extensión aminoterminal y el dominio aminoterminal interactúan con el ADN vírico en el sitio activo del dominio del núcleo catalítico opuesto.
Sitio activo
Los carboxilatos del sitio activo son cadenas laterales de Asp 128, Asp 185, Glu 221.
Un átomo de zinc cada uno está situado cerca de los sitios activos.
- .
- .
Inhibidores de la integrasa
| Nombre | Marca | Empresa | Patente | Notas |
| Raltegravir | Isentress | Merck & Co. | – | también conocido como MK-0518. El isopropilo y el metil-oxadiazol del MK-0518 participan en interacciones hidrofóbicas y de apilamiento con las cadenas laterales de Pro 214 y Tyr 212 para estabilizar este fármaco dentro del sitio activo del intasoma del PFV. Esta forma de interacción de unión al fármaco provoca el desplazamiento del extremo reactivo 3′ del ADN viral del sitio activo del intasoma del VFP. Tras la unión del MK-0518 al sitio activo, el grupo hidroxilo reactivo 3′ se aleja del sitio activo del intasoma del VFP en más de 6 Angstroms. El raltegravir fue aprobado por la FDA el 12 de octubre de 2007 para su uso con otros agentes contra el VIH en el tratamiento de la infección por el VIH en adultos. Es el primer inhibidor de la integrasa aprobado por la FDA. |
| Elvitegravir | – | Gilead Science | – | GS-9137 interactúa con el Pro 214 del intasoma del VFP a través de su base de quinolona y su grupo isopropilo. En fase experimental; comparte la estructura central de los antibióticos de quinolona. Se han completado los estudios de fase II de elvitegravir en personas con experiencia en el tratamiento. Se están realizando estudios de fase III en pacientes con experiencia en el tratamiento. Está en marcha un estudio de fase II de elvitegravir en personas que nunca han tomado terapia antirretroviral. En este estudio también se evaluará un agente potenciador en lugar de Norvir, actualmente llamado GS9350. Elvitegravir es prometedor para los pacientes seropositivos que han tomado otros medicamentos contra el VIH en el pasado. |
| MK-2048 | – | Merck & Co. | – | Un inhibidor de la integrasa de segunda generación, destinado a ser utilizado contra la infección por el VIH. Es superior al primer inhibidor de la integrasa disponible, raltegravir, ya que inhibe la enzima integrasa del VIH 4 veces más. Se está investigando su uso como parte de la profilaxis previa a la exposición (PrEP). |
Ver también Farmacocinética de los inhibidores de la integrasa retroviral.