InterPro contém três entidades principais: proteínas, assinaturas (também referidas como “métodos” ou “modelos”) e entradas. As proteínas no UniProtKB são também as entidades proteicas centrais no InterPro. As informações sobre quais assinaturas correspondem significativamente a estas proteínas são calculadas à medida que as sequências são divulgadas pelo UniProtKB e estes resultados são disponibilizados ao público (ver abaixo). As combinações de assinaturas com proteínas são o que determina como as assinaturas são integradas em conjunto nas entradas InterPro: a sobreposição comparativa dos conjuntos de proteínas combinadas e a localização das combinações das assinaturas nas seqüências são utilizadas como indicadores de correlação. Apenas as assinaturas consideradas de qualidade suficiente são integradas no InterPro. A partir da versão 81.0 (lançada em 21 de agosto de 2020) as entradas InterPro anotadas 73,9% dos resíduos encontrados no UniProtKB com outros 9,2% anotados por assinaturas que estão pendentes de integração.

InterPro também inclui dados para variantes de emendas e as proteínas contidas nos bancos de dados UniParc e UniMES.
Bases de dados de membros do consórcio InterProEditar
As assinaturas do InterPro vêm de 13 “bancos de dados de membros”, que estão listados abaixo.
CATH-Gene3D Descreve famílias de proteínas e arquiteturas de domínio em genomas completos. As famílias proteicas são formadas usando um algoritmo de clustering Markov, seguido de clustering multi-linkage de acordo com a identidade da sequência. O mapeamento dos domínios de estrutura e sequência previstos é feito usando bibliotecas de modelos Markov ocultas que representam os domínios CATH e Pfam. A anotação funcional é fornecida a proteínas de múltiplos recursos. A previsão e análise funcional de arquiteturas de domínios está disponível no site do Gene3D. CDD Conserved Domain Database é um recurso de anotação de proteínas que consiste em uma coleção de modelos de alinhamento de sequências múltiplas anotadas para domínios antigos e proteínas de comprimento total. Estes estão disponíveis como matrizes de pontuação específica de posição (PSSMs) para identificação rápida de domínios conservados em seqüências de proteínas via RPS-BLAST. HAMAP significa Anotação Manual e Automatizada de Proteomas Microbianos de Alta Qualidade. Os perfis HAMAP são criados manualmente por curadores especializados que identificam proteínas que fazem parte de famílias ou subfamílias de proteínas bem conservadas bacterianas, arqueológicas e plastificadas (ou seja, cloroplastos, cianelas, apicoplastos, plastids não fotossintéticos). MobiDB MobiDB é uma base de dados que analiza distúrbios intrínsecos em proteínas. PANTHER PANTHER é uma grande coleção de famílias de proteínas que foram subdivididas em subfamílias funcionalmente relacionadas, utilizando a perícia humana. Estas subfamílias modelam a divergência de funções específicas dentro das famílias protéicas, permitindo uma associação mais precisa com a função (função molecular curada pelo homem e classificações de processos biológicos e diagramas de percurso), bem como a inferência de aminoácidos importantes para a especificidade funcional. Modelos Markov ocultos (HMMs) são construídos para cada família e subfamília para classificação de seqüências proteicas adicionais. Pfam É uma grande coleção de alinhamentos de sequências múltiplas e modelos de Markov ocultos cobrindo muitos domínios e famílias proteicas comuns.
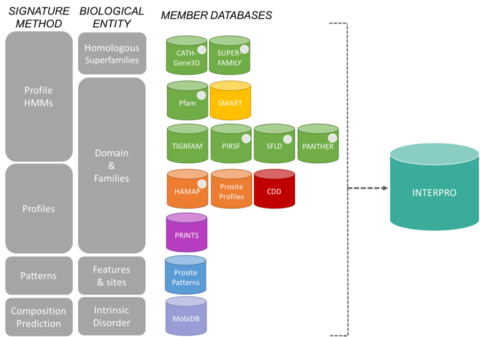
Sistema de classificação de proteínas PIRSF é uma rede com múltiplos níveis de diversidade de sequências desde superfamílias a subfamílias que reflecte a relação evolutiva das proteínas e domínios de comprimento total. A unidade primária de classificação PIRSF é a família homeomórfica, cujos membros são tanto homólogos (evoluíram de um ancestral comum) quanto homeomórficos (compartilhando similaridade de sequência de comprimento total e uma arquitetura de domínio comum). PRINTS PRINTS é um compêndio de impressões digitais de proteínas. Uma impressão digital é um grupo de motivos conservados utilizados para caracterizar uma família de proteínas; o seu poder de diagnóstico é refinado pelo escaneamento iterativo do UniProt. Normalmente os motivos não se sobrepõem, mas são separados ao longo de uma sequência, embora possam ser contíguos no espaço 3D. As impressões digitais podem codificar dobras protéicas e funcionalidades de forma mais flexível e poderosa do que os motivos únicos, sendo toda a sua potência de diagnóstico derivada do contexto mútuo proporcionado pelos vizinhos dos motivos. PROSITE PROSITE é uma base de dados de famílias e domínios protéicos. Consiste em sítios, padrões e perfis biologicamente significativos que ajudam a identificar de forma fiável a que família proteica conhecida (caso exista) pertence uma nova sequência. SMART Simple Modular Architecture Research Tool Permite a identificação e anotação de domínios geneticamente móveis e a análise de arquitecturas de domínios. Mais de 800 famílias de domínios encontrados na sinalização, proteínas extracelulares e associadas à cromatina são detectáveis. Estes domínios são amplamente anotados no que diz respeito às distribuições fléticas, classe funcional, estruturas terciárias e resíduos funcionalmente importantes. SUPERFAMILY SUPERFAMILY é uma biblioteca de modelos Markov de perfil oculto que representam todas as proteínas de estrutura conhecida. A biblioteca é baseada na classificação SCOP de proteínas: cada modelo corresponde a um domínio SCOP e visa representar toda a superfamília SCOP a que o domínio pertence. SUPERFAMILY tem sido utilizado para realizar atribuições estruturais a todos os genomas completamente sequenciados. SFLD Uma classificação hierárquica de enzimas que relaciona características específicas de estrutura sequencial com capacidades químicas específicas. TIGRFAMs TIGRFAMs é uma coleção de famílias de proteínas, apresentando alinhamentos de sequências múltiplas curados, modelos Markov ocultos (HMMs) e anotação, o que fornece uma ferramenta para identificar proteínas funcionalmente relacionadas com base na homologia de sequências. As entradas que são “equivalentes” agrupam proteínas homólogas que são conservadas no que diz respeito à função.
Tipos de dadosEditar
InterPro consiste em sete tipos de dados fornecidos por diferentes membros do consórcio:
| Dados Tipo | Descrição | Contribuindo Bases de Dados |
|---|---|---|
| Entradas InterPro | Estruturais e/ou domínios funcionais de proteínas previstos usando uma ou mais assinaturas | Todos os 13 bancos de dados de membros |
| As assinaturas dos membros do banco de dados | As assinaturas dos membros do banco de dados. Estas incluem assinaturas que são integradas no InterPro, e as que não são | Todos os 13 bancos de dados de membros |
| Proteínas | Sequências de proteínas | UniProtKB (Swiss-Prot e TrEMBL) |
| Proteome | Coleta de proteínas que pertencem a um único organismo | UniProtKB |
| Estrutura | 3-estruturas dimensionais das proteínas | PDBe |
| Taxonomia | Informação taxonómica das proteínas | UniProtKB |
| Set | Grupos de famílias relacionadas com a evolução | Pfam, CDD |

Tipos de entradas InterProEditar
As entradas InterPro podem ser divididas em cinco tipos:
- Superfamília Homóloga: Um grupo de proteínas que compartilham uma origem evolutiva comum, como visto em suas semelhanças estruturais, mesmo que suas seqüências não sejam muito semelhantes. Estas entradas são especificamente fornecidas apenas por duas bases de dados de membros: CATH-Gene3D e SUPERFAMILY.
- Família: Um grupo de proteínas que têm uma origem evolutiva comum determinada através de semelhanças estruturais, funções relacionadas, ou homologia de sequência.
- Domínio: Uma unidade distinta em uma proteína com uma função, estrutura ou sequência particular.
- Repita: Uma sequência de aminoácidos, geralmente não mais que 50 aminoácidos, que tendem a se repetir muitas vezes em uma proteína.
- Local: Uma sequência curta de aminoácidos onde pelo menos um aminoácido é conservado. Estes incluem locais de modificação pós-tradução, locais conservados, locais de ligação e locais ativos.