- Introduction
- Materiały i Metody
- Urządzenia i Systemy Sterowania
- System TPD
- Przygotowanie próbki
- Procedura eksperymentalna
- Próżnia
- Analiza gazów resztkowych
- Chłodzenie
- Adsorpcja
- Desorpcja
- Molekuły adsorbowane
- Wyniki i dyskusja
- Cellulose Desorption/Decomposition Fingerprint
- Spektra TPD
- n-Dekan Desorpcja
- Desorpcja deuterowanego metanolu
- Wnioski
- Wkład Autora
- Oświadczenie o konflikcie interesów
- Podziękowania
- Supplementary Material
Introduction
Dyskusja na temat korzyści vs. kosztów środowiskowych materiałów opakowaniowych jest stała. W dobie stale rosnącej świadomości ekologicznej, klienci często nagradzają „ekologiczne” postępowanie, a papier bezpiecznie zajmuje czołowe miejsce w tej kwestii.
Jednakże, aby papier mógł być stosowany jako materiał opakowaniowy do żywności, musi spełniać szereg wymagań. Z jednej strony cząsteczki aromatu muszą pozostać wewnątrz żywności, a z drugiej strony opakowanie jest zakładane w celu ochrony żywności przed wpływami środowiska, takimi jak tlen lub cząsteczki organiczne, które mogłyby spowodować dojrzewanie lub wytworzyć zgniły smak. Ponieważ papier jest materiałem porowatym, kluczową cechą jest transport molekuł przez porowatą strukturę, co pozwala uzyskać wgląd i kontrolę nad wyżej wymienionymi kwestiami. Jednym z pytań, na które należy odpowiedzieć w tym kontekście, jest stopień, w jakim cząsteczki adsorbują się na ściankach porów. To sprawia, że badania adsorpcji molekuł na powierzchniach celulozowych są tym bardziej istotne.
Badania literaturowe ujawniają znaczną liczbę badań nad materiałami celulozowymi w stanie mokrym w celu usuwania zanieczyszczeń z wody (patrz np., Voisin et al., 2017). Intensywnie badano również adsorpcję polielektrolitów na masach celulozowych (zob. np. Li i in., 2003; Neumann i in., 2018). Adsorpcja mniejszych cząsteczek przyciąga mniej uwagi, co zostało udowodnione w niedawnym przeglądzie (Lombardo i Thielemans, 2019). Adsorpcja związków aromatycznych na celulozie była badana przez Pereza i wsp. (2004). Adsorpcja benzofenonu była badana przez Mazeau i Vergelati (2002), a adsorpcja czerwieni Kongo przez Mazeau i Wyszomirskiego (2012). Adsorpcja naftaliny i pirenu była badana przez Tozuka i wsp. (1998, 2002). Adsorpcja mocznika została zbadana przez Chen et al. (2017), aby wymienić tylko kilka przykładów. Artykuły te nie są jednak istotne dla dziedziny opakowań do żywności. W związku z tym badamy adsorpcję małych cząsteczek z fazy gazowej na powierzchniach celulozowych. W tym kontekście dynamiczna sorpcja pary wodnej jest szeroko badana (patrz np. Xie et al., 2011). Zazwyczaj takie badania odbywają się w warunkach równowagi. Kiedy adsorpcja małych cząsteczek zachodzi w opakowaniach papierowych, jest mało prawdopodobne, że warunki równowagi mają zastosowanie. Proponujemy raczej zbadanie adsorpcji małych cząsteczek w warunkach wysokiej próżni, dalekich od warunków równowagi. Niestety, takie pomiary są niemożliwe przy użyciu próbek papierowych, ponieważ papier nie może być ogrzewany bezpośrednio w komorze próżniowej. Z tego powodu musimy zastosować system modelowy, w naszym przypadku cienkie folie celulozowe. Pomimo faktu, że takie cienkie folie celulozowe są produkowane z celulozy II zamiast z celulozy I, która występuje w papierze, wykazano, że folie te są użytecznym zastępczym systemem modelowym dla papieru (Rohm i in., 2014). Cienkie folie celulozowe były również wcześniej wykorzystywane jako podłoża do adsorpcji wody (patrz np. Niinivaara i in., 2015, 2016; Hakalahti i in., 2017) oraz adsorpcji cukru (Hoja i in., 2014).
W niniejszym opracowaniu przedstawiamy pierwsze oparte na wysokiej próżni badania adsorpcji i programowanej temperaturowo spektroskopii desorpcyjnej n-dekanu i deuterowanego metanolu na cienkich foliach celulozowych.
Materiały i Metody
Urządzenia i Systemy Sterowania
– Pompa łopatkowa:
Leybold TRIVAC D 25 B
– Turbo pompa molekularna &system sterowania:
Oerlikon Leybold Vacuum, Turbovac typ TMP151 & Turbo.Drive TD20 classic
– Kwadrupolowy spektrometr mas & oprogramowanie sterujące:
Balzers Prisma typ QME 200 (CF DN40) & Quadstar 32-Bit
– zasilacz:
HP DC Power Supply System 6261B
– Układ regulacji temperatury
Kontroler PID (Proportional-Integral-Derivative) w National Instruments LabVIEW 7.0
– Układ sterowania manometrem Bayarda-Alperta &:
Leybold AG IE 414 (CF DN40) & IONIVAC IM 520
– Ręczny zawór kątowy
Balzers EVA 025 H (KF DN25)
– Zawór wlotowy gazu
Pfeiffer Vacuum Gas Dosing Valve EVN 116 (KF DN16)
– Złącze metal-szkło skazane na zagładę:
GMA-KF16-KGD-152-TD13 (KF DN16).
System TPD
Główną komorą próżniową jest sześciodrożne przejście ze stali nierdzewnej. System pompujący składa się z pompy molekularnej turbo, przymocowanej do dolnej części komory głównej (patrz rysunek 1), a następnie z obrotowej pompy łopatkowej, nie pokazanej na rysunku 1. Kwadrupolowy spektrometr masowy i miernik Bayarda-Alperta są zamontowane po lewej i po prawej stronie komory. Zawór wlotowy gazu jest przymocowany z tyłu i połączony ze szklanym pojemnikiem zawierającym ciekłe cząsteczki organiczne (patrz rysunek 1 widok z tyłu).
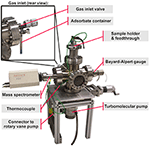
Rysunek 1. System TPD, przegląd.
Górna strona sześciodrożnego skrzyżowania jest przedłużona przez adapter do pokrywy kołnierzowej DN63 CF, która jest mocowana przez szybki łańcuch zaciskowy CF zapewniający szybszą wymianę próbki. Pokrywa kołnierzowa oferuje dwa elektryczne i jeden termoparowy przepust, jak również jeden otwór do miedzianego korpusu palca chłodzącego. Palec chłodzący, wydrążony cylindryczny element miedziany, jest zamontowany na wewnętrznej stronie pokrywy kołnierzowej, rozciągając się w dół do środka komory, łącząc się ze spłaszczonym elementem końcowym, zwanym później „uchwytem próbki” (Rysunek 2). Do tej platformy przymocowane są dwie ławy do przechowywania próbek, odizolowane za pomocą przekładek z tlenku aluminium, które zapewniają izolację elektryczną i przewodzenie ciepła.

Rysunek 2. Uchwyt próbki systemu TPD, widok z góry i z boku.
Podłożem próbki jest płytka ze stali nierdzewnej o wymiarach 1 × 1 cm z dwoma drutami Ta do ogrzewania oporowego i termoparą typu K, zgrzaną punktowo do jej tylnej strony. Podłoże jest montowane poprzez zaciśnięcie drutów grzejnych w ławkach mocujących próbki, które są połączone z przepustem elektrycznym za pomocą kabli miedzianych.
W środkowej części, palec chłodzący posiada również dwa minizłącza termoparowe z tlenku aluminium, które są ze sobą połączone. Podczas gdy górne mini złącze jest na stałe podłączone do przepustu termopary w pokrywie kołnierza, dolne złącze działa jako płytka zaciskowa dla przewodów termopary pochodzących z tylnej strony próbki (patrz rysunek 2).
W celu zwiększenia przepływu gazu do powierzchni próbki, rura wlotu gazu, która jest podłączona do zaworu wlotu gazu (patrz rysunek 3), skierowana jest na powierzchnię próbki pod kątem około 35° i kończy się w odległości około 3 cm od powierzchni próbki. Spektrometr masowy jest umieszczony dokładnie naprzeciwko powierzchni próbki w tej samej odległości.

Rysunek 3. Geometria wewnętrzna komory próżniowej.
Przygotowanie próbki
Próbkę przygotowuje się według tej samej standardowej procedury, którą zaproponowali Kontturi et al. (2003) oraz Rohm et al. (2014), przy czym podłoże pokrywa się cienką warstwą celulozy. Celuloza, o której mowa, to trimetylosililoceluloza (TMSC) o stopniu podstawienia około 2,8 (Thüringisches Institut für Textil-und Kunstoff-Forschung e.V. (TITK), Niemcy), rozpuszczona w toluenie o stężeniu około 1 mg/ml. Otrzymany roztwór nanosi się kroplami na wypolerowaną metalową powierzchnię próbki i suszy na powietrzu. Następnie, w szalce Petriego umieszcza się kroplę 10% HCl, a próbkę umieszcza się tuż obok kropli HCl. Naczynie wraz z próbką jest następnie przykrywane folią aluminiową i pozostawiane do reakcji na 15 min.
Według Konturri i Djak procedura ta prowadzi do uzyskania w pełni zregenerowanej błony celulozowej o grubości około 50 nm (Kontturi et al., 2003; Djak et al., 2011). Pomiary FTIR folii nie wykazały żadnych drgań indukowanych trimetylosilem w foliach z czystej celulozy. Podczas desorpcji termicznej znaleziono jednak masę 73, która może odpowiadać fragmentowi trimetylosililowemu. Ilość resztkowego TMSC leży zatem poniżej granicy wykrywalności FTIR (~1 × 1013 cząsteczek) i powyżej granicy wykrywalności spektroskopii masowej. Grubość warstwy kilku innych próbek również zmierzono za pomocą AFM. Później te próbki AFM nie były już używane do eksperymentów desorpcyjnych, aby uniknąć wpływu rys spowodowanych pomiarami grubości AFM na zachowanie desorpcyjne.
Filmy są całkowicie amorficzne; dlatego nie spodziewamy się zobaczyć w nich struktury porowatej. Podczas badań AFM takich filmów (Ganser i in., 2014, 2016; Rohm i in., 2014) nie znaleziono żadnych oznak porów. Wskazuje to, że gęstość powinna być zbliżona do gęstości zregenerowanej celulozy. Niemniej jednak, porowatość i gęstość folii nie były mierzone w ramach tego badania.
Procedura eksperymentalna
Próżnia
Próbka jest montowana w ławkach do trzymania próbek i podłączana jest termopara (patrz wyżej). Uchwyt próbki jest wkładany do komory próżniowej i rozpoczyna się monitorowanie temperatury. Pokrywę kołnierza mocuje się za pomocą łańcucha szybkiego zacisku CF, a komorę próżniową opróżnia się do ciśnienia podstawowego wynoszącego około 5 × 10-9 mbar. Dzięki żarnikom spektrometru masowego i miernika Bayarda-Alperta temperatura próbki wewnątrz komory próżniowej utrzymuje się na poziomie 55°C, podczas gdy wykonywana jest ewakuacja.
Analiza gazów resztkowych
Analizę gazów resztkowych przeprowadza się rejestrując widmo masowe od masy 1 do masy 100 (takie widmo masowe przedstawiono w informacjach dodatkowych (SI Rysunek 1) w celu oceny warunków początkowych i jakości próżni.
Chłodzenie
Ciekły azot (N2(l)) jest teraz wielokrotnie napełniany do palca chłodzącego. Po osiągnięciu zadanej temperatury podstawowej próbki (-80°C), jest ona utrzymywana przez umiarkowane, sterowane PID (Proportional-Integral-Derivative), ogrzewanie oporowe i dalsze chłodzenie N2(l) w razie potrzeby.
Adsorpcja
Zawór wlotowy gazu jest teraz ostrożnie otwierany. Ze względu na ciśnienie pary w temperaturze pokojowej cząsteczek organicznych wewnątrz pojemnika z adsorbatem, para jest dozowana do komory próżniowej. Podczas adsorpcji próbka jest utrzymywana w temperaturze -80°C. Zawór jest regulowany tak długo, aż napływ gazowych cząsteczek organicznych i pompowanie przez turbo pompę molekularną wyrówna się do ciśnienia stanu ustalonego 5 × 10-6 mbar. Ciśnienie to jest następnie utrzymywane przez określony czas adsorpcji wynoszący 6 minut. Po tym czasie zawór wlotowy gazu jest szybko zamykany, a ciśnienie w komorze powraca do ciśnienia bazowego.
Desorpcja
Osiągnięcie ciśnienia bazowego trwa około 1 min, a proces desorpcji termicznej rozpoczyna się od temperatury -80°C. Regulator PID podgrzewa próbkę z szybkością grzania 1°C/s do górnej granicy temperatury 600°C. Podczas desorpcji sygnały intensywności zadanych mas są w sposób ciągły rejestrowane w czasie przez spektrometr masowy. Równocześnie rejestrowana jest temperatura próbki w funkcji czasu. Intensywność każdej masy jest następnie repotowana jako funkcja temperatury. Jest to dość typowa konfiguracja dla pomiarów TPD (patrz np. Hohenegger et al., 1998).
Ponieważ tylko ograniczona liczba natężeń mas może być zarejestrowana w rzędzie, każda masa reprezentuje charakterystyczny fragment uczestniczącej cząsteczki: masa 57 dla n-dekanu, masa 34 dla deuterowanego metanolu (D3COD), masa 44 dla CO2 i masa 18 dla H2O. Podczas gdy dwie pierwsze masy są charakterystyczne dla zaadsorbowanych gatunków, dwie ostatnie masy są charakterystyczne dla desorpcji celulozy.
Molekuły adsorbowane
Cząsteczki aromatyczne są zazwyczaj cząsteczkami organicznymi o dość wysokim ciśnieniu pary. Można spotkać zarówno polarne, jak i niepolarne cząsteczki zapachowe. Aby uzyskać pierwsze wyobrażenie o zachowaniu adsorpcyjnym takich cząsteczek na cienkiej warstwie celulozy, użyliśmy n-dekanu jako cząsteczki niepolarnej i deuterowanego metanolu (D3COD) jako cząsteczki polarnej. Obie molekuły otrzymano w jakości p.a. od firmy Sigma Aldrich (deuterowany metanol ≥99%, czystość izotopowa ≥99.8 atom % D, n-dekan ≥99.5%) i oczyszczono in situ poprzez kilka cykli zamrażania i rozmrażania. Masa cząsteczkowa n-dekanu wynosi 142 amu, a ciśnienie pary w temperaturze 30°C wynosi 3,17 hPa, co okazało się wystarczające do adsorpcji. Ponieważ zastosowany kwadrupolowy spektrometr mas ma maksymalną masę 100 amu, musieliśmy szukać masy 57 dla n-dekanu. W końcu okazało się, że jest to również najwyższy pik w widmie masowym n-dekanu. Masa 57 ma również raczej niską intensywność w widmie masowym desorbującej folii celulozowej. Widmo masowe n-dekanu można znaleźć w informacjach uzupełniających (SI Rysunek 2). Widmo masowe n-dekanu pozostało niezmienione w czasie i nie wykazuje żadnych dowodów na zanieczyszczenie niższymi węglowodorami. Niemniej jednak, nie można całkowicie wykluczyć śladów zanieczyszczenia nonanem i oktanem.
Masa cząsteczkowa deuterowanego metanolu wynosi 36 amu. Najwyższy pik w widmie masowym deuterowanego metanolu znajduje się przy 34 amu. W naszych pomiarach TPD użyliśmy tej masy jako wskaźnika, ponieważ nie koliduje ona ani z widmem masowym desorbującej folii celulozowej, ani z n-dekanem. Widmo masowe deuterowanego metanolu podczas naświetlania można znaleźć również w informacjach dodatkowych (SI Rysunek 3). Ciśnienie pary deuterowanego metanolu w 20°C wynosi 129 hPa.
Wyniki i dyskusja
Naświetlone cząsteczki nie tylko adsorbują się na folii celulozowej, ponieważ adsorpcja odbywa się poprzez fazę gazową. Dlatego też przeprowadzono serię ślepych eksperymentów, aby upewnić się, że stwierdzone efekty pochodzą wyłącznie z folii celulozowych. W ślepym reżimie testowano czyste folie podłoża z adsorpcją i bez adsorpcji n-dekanu i deuterowanego metanolu. Te trzy eksperymenty wyraźnie pokazują piki desorpcji różniące się od tych przedstawionych tutaj. Bez wcześniejszej adsorpcji gazu na błonach celulozowych można znaleźć (patrz Rysunek 4) kilka fragmentów desorbujących w tym samym zakresie temperatur, co pokazują poniższe widma TPD.
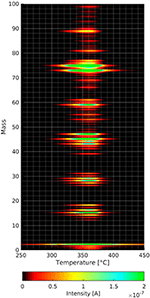
Rysunek 4. Widma masowe w funkcji temperatury rozkładu i desorpcji folii celulozowej. Intensywności poszczególnych mas są powiązane z kodem kolorystycznym.
Cellulose Desorption/Decomposition Fingerprint
Rysunek 4 przedstawia wynik eksperymentu TPD z folią celulozową bez wcześniejszej adsorpcji gazu. Na trójwymiarowym wykresie można zauważyć, że maksymalna intensywność fragmentów desorpcji celulozy występuje w temperaturze około 360°C. Jest to niższa temperatura niż dla innych widm TPD pokazanych później. Szybkość ogrzewania we wspomnianym eksperymencie wynosi tylko 0.1°C/s, w przeciwieństwie do 1°C/s we wszystkich pozostałych eksperymentach. Niższa szybkość ogrzewania stała się konieczna, ponieważ uzyskanie pełnego widma masowego od 0 do 100 amu trwa znacznie dłużej niż w przypadku 4 pojedynczych mas w innych eksperymentach pokazanych później.
Na rysunku 4 można zauważyć, że zarówno masa 18 jak i masa 44 desorbują podczas rozkładu i desorpcji celulozy. Najbardziej intensywne piki desorpcji występują przy masach 73 i 75 amu. Podczas gdy pik przy masie 73 pasuje do grupy trimetylosililowej, nie można wyjaśnić masy 75 za pomocą tej grupy, ponieważ izotop Si o masie 30 występuje tylko w około 3%. W tym kontekście, masa 75 może również pochodzić z fragmentacji większych gatunków, których nie jesteśmy w stanie wykryć ze względu na ograniczenie masowe (masa 100) zastosowanego spektrometru masowego. Można zauważyć kilka skupisk mas o odległości około 10 amu, co jest częstą cechą widm masowych cząsteczek organicznych. Niemniej jednak, należy dokładnie rozważyć, które masy śledzić podczas wykonywania eksperymentów termicznej desorpcji z błon celulozowych. Pomaga to odróżnić zaadsorbowany gatunek od rozkładającej się/desorbującej warstwy celulozy. Wybraliśmy masę 57 dla n-dekanu, ponieważ ten pik jest najmniejszym z pików celulozy, który może być użyty dla n-dekanu. Deuterowany metanol został użyty, ponieważ jego pik o najwyższej masie 34 amu nie jest wykrywany w widmie desorpcji filmu celulozowego.
Zauważyliśmy również we wstępnych eksperymentach, że ogrzewanie do 150°C prowadziło do wyraźnej zmiany w cienkich filmach celulozowych, pokazanej przez znaczącą zmianę początku widma TPD po powtórzeniu pomiarów na tym samym filmie (patrz SI Rysunek 4). W niniejszej pracy przedstawiamy tylko dane uzyskane z dziewiczej cienkiej folii celulozowej. Desorpcja folii celulozowych jest zakończona w temperaturze około 600°C. Z czasem stwierdziliśmy nagromadzenie pozostałości węgla na podłożu, który został usunięty przez polerowanie powierzchni przed utworzeniem filmu.
Spektra TPD
Interpretacja widm TPD w Surface Science jest zgodna z zasadą równowagi mikroskopowej. Gatunek adsorbujący będzie zatem podążał tą samą drogą podczas adsorpcji i desorpcji. Jako wniosek odwrotny można się czegoś dowiedzieć o procesie adsorpcji z eksperymentów desorpcji (Somorjai, 1981). Wykazano, że kształt piku desorpcyjnego z eksperymentu TPD ma charakterystyczny kształt dla desorpcji z wielowarstwy zarówno dla desorpcji molekularnej jak i dla adsorpcji dysocjacyjnej (Somorjai, 1981). Redhead zaproponował uproszczoną metodę obliczania energii adsorpcji na podstawie temperatury w maksimum piku widma desorpcji termicznej (Redhead, 1962). W niniejszej pracy podajemy energie adsorpcji dla zaadsorbowanych gatunków obliczone zgodnie ze wzorem Redheada. W każdym razie należy pamiętać, że metoda ta była pierwotnie przeznaczona do adsorpcji prostych molekuł, takich jak CO lub H2. Nie zbadano jeszcze, w jakich warunkach metoda Redheada ma zastosowanie do cząsteczek organicznych. Ostatecznie, wartości te dają przynajmniej pojęcie o energii adsorpcji. Różne energie adsorpcji są powszechnie związane z różnymi miejscami adsorpcji na powierzchni. Ten związek jest używany w całym tym artykule.
n-Dekan Desorpcja
Asymetryczny kształt piku ze stromą krawędzią spływu piku desorpcji, jak pokazano na rysunku 5, jest wyraźnym znakiem, że n-dekan desorbuje z wielowarstwy, która była adsorbowana w temperaturze -80°C. Wielowarstwy desorbują pomiędzy około -70 i -52°C z energią adsorpcji około 60 kJ/mol. Wartość ta mieści się pomiędzy ciepłem parowania 56 kJ/mol w -15°C (Carruth i Kobayashi, 1973) i 77 kJ/mol otrzymanym przez ekstrapolację danych z Majer i Scoboda (1985) do -70°C. Sugeruje to, że ten pik desorpcji pochodzi z wielowarstwy na powierzchni. W tym samym zakresie temperatur można również zaobserwować pewną desorpcję CO2, która najprawdopodobniej pochodzi z adsorpcji z gazu resztkowego podczas chłodzenia i adsorpcji n-dekanu. Bliższe spojrzenie na ten sam zakres temperatur pokazuje również pik desorpcji wody (patrz Rysunek 6).
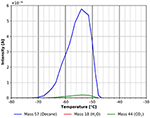
Rysunek 5. Desorpcja n-dekanu (masa 57, kolor niebieski), dwutlenku węgla (masa 44, kolor zielony) i wody (masa 18, kolor czerwony) w temperaturze od -80 do -30°C.
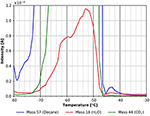
Rysunek 6. Powiększenie widma TPD pokazanego na Rysunku 5. n-dekan (masa 57, niebieski), dwutlenek węgla (masa 44, zielony) i woda (masa 18, czerwony).
Pik desorpcji wody widoczny w zakresie temperatur pomiędzy -80 i około -52°C najprawdopodobniej jest spowodowany adsorpcją wody z gazu resztkowego podczas chłodzenia i ekspozycji na gaz. Może on również częściowo wynikać z obecności wody resztkowej w warstwie celulozy. Aby zminimalizować ten ostatni efekt, przedstawione tu eksperymenty rozpoczęto dopiero po osiągnięciu ciśnienia tła wynoszącego około 5 ×10-9 mbar. Wpływ ciśnienia tła przed adsorpcją jest pokazany w informacjach dodatkowych (SI Rysunek 5). Rysunek 6 pokazuje również inną cechę desorpcji z n-dekanu i wody z maksimum przy około -43°C. Nawet jeśli ten pik jest mały, proszę zauważyć, że wszystkie liczby pochodzą bezpośrednio z surowych danych bez żadnego wygładzania. Z tego powodu wszystkie piki omawiane w tej pracy wykazują dobry stosunek sygnału do szumu i wszystkie omawiane cechy zostały eksperymentalnie odtworzone kilka razy.
Cecha ta jest pokazana bardziej szczegółowo na Rysunku 7. Pik desorpcji przy około -43°C wyraźnie sugeruje silniej związany gatunek n-dekanu. Używając wzoru Redheada odpowiada to energii wiązania około 70 kJ/mol. Mały pik CO2 i wody jest nadal wyraźnie widoczny na Rysunku 7, co wskazuje, że te gatunki znajdują podobne miejsca adsorpcji na powierzchni celulozy.
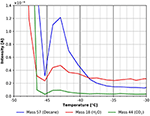
Rysunek 7. Powiększenie widma TPD pokazanego na Rysunku 6. n-dekan (masa 57, niebieski), dwutlenek węgla (masa 44, zielony) i woda (masa 18, czerwony).
Przechodząc do wyższych temperatur można znaleźć małą i szeroką cechę desorpcji n-dekanu pomiędzy około -40 i +14°C (patrz Rysunek 8) odpowiadającą energii adsorpcji około 160 kJ/mol. Cecha ta nie występuje w śladzie desorpcji CO2 i nie jest wyraźnie widoczna w śladzie desorpcji wody. Przyczyną większego rozproszenia w danych na rysunku 8 mogła być szybkość ogrzewania, która nie była idealnie liniowa w tym zakresie temperatur.
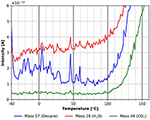
Rysunek 8. Desorpcja n-dekanu (masa 57, niebieski), dwutlenku węgla (masa 44, zielony) i wody (masa 18, czerwony) w zakresie temperatur od -40 do +160°C.
Rysunek 8 pokazuje również, że wszystkie trzy gatunki zaczynają desorbować w różnych temperaturach początkowych wynoszących około 80°C dla wody, 95°C dla n-dekanu i 100°C dla CO2. Jest to również początek desorpcji/degradacji warstwy celulozy, jak można zobaczyć na Rysunku 8. Inną silną wskazówkę, że film celulozowy ulega znaczącym zmianom podczas ogrzewania, można dostrzec w różnicy między początkiem desorpcji po powtarzających się cyklach adsorpcji i desorpcji bez przygotowania nowego filmu, pokazanych w informacjach dodatkowych (SI Rysunek 4).
Największym pikiem desorpcji na Rysunku 9 jest pik CO2, po którym następuje pik wody. Zaskakująco, nadal widoczna jest znaczna desorpcja n-dekanu. Pik ten może również pochodzić częściowo z rozkładu/desorpcji warstwy celulozy. Niestety, te dwie możliwości nie mogą być w tej chwili odróżnione od siebie. Jak wspomniano powyżej, na rysunku 4 widoczny jest fragment przy masie 57. Prawdopodobnie istnieje tam 4 miejsce adsorpcyjne dla n-dekanu o energii wiązania około 180 kJ/mol. Degradacja celulozy zachodzi również w mniej więcej tym samym zakresie temperatur. Wyniki te pokazują, że cząsteczki niepolarne łatwo adsorbują się na powierzchniach celulozy w temperaturze pokojowej i wyższej.
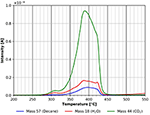
Rysunek 9. Desorpcja n-dekanu (masa 57, kolor niebieski), dwutlenku węgla (masa 44, kolor zielony) i wody (masa 18, kolor czerwony) w zakresie temperatur od 200 do 550°C.
Porównanie rysunków 9 i 10 pokazuje, że na końcową desorpcję wszystkich gatunków mają wpływ zaadsorbowane cząsteczki. W przypadku adsorpcji n-dekanu na Rysunku 9 można zaobserwować trzy różne piki desorpcji dla wszystkich trzech badanych gatunków. Dla deuterowanego metanolu obserwuje się tylko jeden pik w tym samym obszarze temperatury (Rysunek 10). Sugeruje to, że zaadsorbowane gatunki mają wpływ na rozkład i desorpcję celulozy.
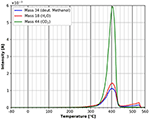
Rysunek 10. Desorpcja deuterowanego metanolu (masa 34, niebieski), ditlenku węgla (masa 44, zielony) i wody (masa 18, czerwony) w zakresie temperatur od -80 do 560°C.
Desorpcja deuterowanego metanolu
Rysunek 10 przedstawia widmo TPD z deuterowanego metanolu w całym zakresie temperatur. Widać wyraźnie, że piki niskotemperaturowe są w tym przypadku nieobecne. Jest to silny wskaźnik, że polarny deuterowany metanol jest silniej związany podczas adsorpcji niż n-dekan. Niemniej jednak, energia adsorpcji zgodnie z równaniem Redheada wynosi również około 180 kJ/mol. Oczywiście należy pamiętać, że pik desorpcji masy 57 w przypadku n-dekanu pokrywa się z pikiem masy 57 pochodzącym z desorpcji/dekompozycji celulozy, co nie ma miejsca w przypadku desorpcji deuterowanego metanolu. Cała desorpcja masy 57 zachodzi w niższych temperaturach i dlatego niższe energie adsorpcji są zupełnie nieobecne w przypadku deuterowanego metanolu. Dodatkowo, wydaje się, że istnieje wyraźny wpływ głównej adsorbowanej cząsteczki na adsorpcję wody i CO2 z gazu resztkowego. Wpływ deuterowanego metanolu na samą błonę celulozową można zaobserwować w głównym piku przy 400°C. W przeciwieństwie do przypadku n-dekanu, po adsorpcji deuterowanego metanolu nie występują żadne ramiona w piku degradacji/desorpcji. Po adsorpcji deuterowanego metanolu, brak jest małego piku występującego w około 310°C po adsorpcji n-dekanu, jak również ramion w głównym piku desorpcji. Możemy również wykryć niewielką dodatkową desorpcję wszystkich trzech gatunków w temperaturze około 550°C, która nie była widoczna po adsorpcji n-dekanu. Niestety, w tej temperaturze nie można całkowicie wykluczyć artefaktu miedzianego uchwytu próbki.
Wyniki przedstawione na rysunkach 9, 10 pokazują, że adsorpcja polarnych cząsteczek ma silny wpływ na cienkie błony celulozowe, ponieważ widma desorpcji są znacząco różne.
Rysunek 11 pokazuje bardziej szczegółowo punkt początkowy widm TPD po adsorpcji deuterowanego metanolu. Desorpcja wody rozpoczyna się w temperaturze około -60°C. Fluktuacje desorpcji wody w tym zakresie temperatur są najprawdopodobniej spowodowane małymi fluktuacjami w liniowej szybkości ogrzewania. Co ciekawe, desorpcja deuterowanego metanolu po raz pierwszy rozpoczyna się w temperaturze około +40°C. Ta różnica w punktach początkowych desorpcji pomiędzy wodą i deuterowanym metanolem może być przypisana różnym miejscom adsorpcji. Wydaje się, że metanol jest silniej związany z warstwą celulozy niż woda. Desorpcja CO2 rozpoczyna się w temperaturze około +60°C, co oznacza początek rozkładu celulozy i desorpcji.
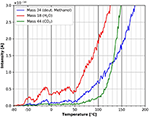
Rysunek 11. Zbliżenie desorpcji deuterowanego metanolu (masa 34, niebieski), dwutlenku węgla (masa 44, zielony) i wody (masa 18, czerwony) w zakresie temperatur od -80 do 200°C.
Wnioski
W niniejszej pracy badano adsorpcję i desorpcję cząsteczek organicznych na cienkich błonach celulozowych z wykorzystaniem programowanej temperaturowo desorpcji. Wykazano, że adsorpcja wody i CO2 z gazu resztkowego jest również zależna od zaadsorbowanych cząsteczek. Wszystkie gatunki mają wyraźny wpływ na proces rozkładu/desorpcji folii celulozowych. Z przedstawionych tu wyników jasno wynika, że zarówno cząsteczki niepolarne jak i polarne łatwo adsorbują się na powierzchni celulozy, przy czym cząsteczka polarna wykazuje wyższą energię wiązania.
Deuterowany metanol wykazuje pik desorpcji w temperaturze około 400°C i w tym samym zakresie temperatur zachodzi desorpcja/desorpcja celulozy. Ponieważ zaadsorbowane gatunki desorbują w tym samym zakresie co film celulozowy, energia adsorpcji może być w rzeczywistości wyższa niż przybliżone 180 kJ/mol otrzymane z równania Redheada.
W przyszłości planujemy zbadać inne molekuły, jak również możliwy wpływ grubości filmu celulozowego na adsorpcję i desorpcję molekuł organicznych.
Wkład Autora
EH zaplanował i zbudował komorę próżniową oraz przeprowadził eksperymenty. VH przeprowadził wstępne eksperymenty. JA przeprowadził wstępne eksperymenty. RS miał pomysł na badania, ustawienie komory próżniowej i nadzorował studentów.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Ta praca została wsparta przez Christian Doppler Society, Austria. The financial support of the Austrian Federal Ministry for Digital and Economic Affairs and the National Foundation for Research, Technology and Development, Austria is gratefully acknowledged.
Supplementary Material
Materiały uzupełniające do tego artykułu można znaleźć online pod adresem: https://www.frontiersin.org/articles/10.3389/fmats.2019.00178/full#supplementary-material
Carruth, G. F., and Kobayashi, R. (1973). Vapor pressure of normal paraffins ethane through n-decane from their triple points to about 10 mm mercury. J Chem Eng Data. 18, 115-126. doi: 10.1021/je60057a009
CrossRef Full Text | Google Scholar
Chen, P., Nishiyama, Y., Wohlert, J., Lu, A., Mazeau, K., and Ismail, A. E. (2017). Translational entropy and dispersion energy jointly drive the adsorption of urea to cellulose. J. Phys. Chem. B 121, 2244-2251. doi: 10.1021/acs.jpcb.6b11914
PubMed Abstract | CrossRef Full Text | Google Scholar
Djak, M., Gilli, E., Kontturi, E., and Schennach, R. (2011). Thickness dependence of reflection-absorption infrared spectra of supported thin polymer films. Macromolecules 44, 1775-1778. doi: 10.1021/ma102905v
CrossRef Full Text | Google Scholar
Ganser, C., Hirn, U., Rohm, S., Schennach, R., and Teichert, C. (2014). AFM nanoindentation of pulp fibers and thin cellulose films at varying relative humidity. Holzforschung 68, 53-60. doi: 10.1515/hf-2013-0014
CrossRef Full Text | Google Scholar
Ganser, C., Niegelhell, K., Czibula, C., Chemelli, A., Teichert, C., Schennach, R., et al. (2016). Efekty topograficzne w eksperymentach mapowania siłowego AFM na cienkich foliach celulozowych dekorowanych ksylanem. Holzforschung 70, 1115-1123. doi: 10.1515/hf-2016-0023
CrossRef Full Text | Google Scholar
Hakalahti, M., Faustini, M., Boissiere, C., Kontturi, E., and Tammelin, T. (2017). Interfacjalne mechanizmy sorpcji pary wodnej do filmów z nanofibryli celulozy ujawnione przez modele ilościowe. Biomacromolecules 18, 2951-2958. doi: 10.1021/acs.biomac.7b00890
PubMed Abstract | CrossRef Full Text | Google Scholar
Hohenegger, M., Bechtold, E., and Schennach, R. (1998). Coadsorption of oxygen and chlorine on Pt(111). Surf. Sci. 412/413, 184-191.
Google Scholar
Hoja, J., Maurer, R. J., and Sax, A. F. (2014). Adsorpcja glukozy, celobiozy i celotetraozy na modelowych powierzchniach celulozy. J Phys. Chemistry B 118, 9017-9027. doi: 10.1021/jp5025685
PubMed Abstract | CrossRef Full Text | Google Scholar
Kontturi, E., Thüne, P. C., and Niemantsverdriet, J. W. (2003). Novel method for preparing cellulose model surfaces by spin coating. Polymer 44, 3621-3625. doi: 10.1016/S0032-3861(03)00283-0
CrossRef Full Text | Google Scholar
Li, H., Du, Y., and Xu, Y. (2003). Adsorpcja i kompleksowanie chitozanowych dodatków do mokrych końcówek w systemach papierniczych. J Appl Polymer Sci. 91, 2642-2648. doi: 10.1002/app.13444
CrossRef Full Text | Google Scholar
Lombardo, S., and Thielemans, W. (2019). Thermodynamics of adsorption on nanocellulose surfaces. Cellulose 26, 249-279. doi: 10.1007/s10570-018-02239-2
CrossRef Full Text | Google Scholar
Majer, V., and Scoboda, V. (1985). Enthalpies of vaporization of organic compounds: a critical review and data compilation. Blackwell Sci. Publications. 1985:300.
Mazeau, K., and Vergelati, C. (2002). Atomistic Modeling of the Adsorption of benzophenone onto cellulosic surfaces. Langmuir 18, 1919-1927. doi: 10.1021/la010792q
CrossRef Full Text | Google Scholar
Mazeau, K., and Wyszomirski, M. (2012). Modelling of Congo red adsorption on the hydrophobic surface of cellulose using molecular dynamics. Cellulose 19, 1495-1506. doi: 10.1007/s10570-012-9757-6
CrossRef Full Text | Google Scholar
Neumann, R. D., Berg, J. M., and Claesson, P. M. (2018). Bezpośredni pomiar sił powierzchniowych w systemach wytwarzania papieru i powlekania papieru. Nordic Pulp Paper Res. J. 8, 96-104. doi: 10.3183/npprj-1993-08-01-p096-104
CrossRef Full Text
Niinivaara, E., Faustini, M., Tammelin, T., and Kontturi, E. (2015). Water vapor uptake of ultrathin films of biologically derived nanocrystals: quantitative assessment with quartz crystal microbalance and spectroscopic ellipsometry. Langmuir 31, 12170-12176. doi: 10.1021/acs.langmuir.5b01763
PubMed Abstract | CrossRef Full Text | Google Scholar
Niinivaara, E., Faustini, M., Tammelin, T., and Kontturi, E. (2016). Mimicking the humidity response of the plant cell wall by using two-dimensional systems: the critical role of amorphous and crystalline polysaccharides. Langmuir 32, 2032-2040. doi: 10.1021/acs.langmuir.5b04264
PubMed Abstract | CrossRef Full Text | Google Scholar
Perez, D. D. S., Ruggiero, R., Morais, L. C., Machado, A. E. H., and Mazeau, K. (2004). Theoretical and experimental studies on the adsorption of aromatic compounds on cellulose. Langmuir 20, 3151-3158. doi: 10.1021/la0357817
CrossRef Full Text | Google Scholar
Redhead, P. A. (1962). Termiczna desorpcja gazów. Vacuum 12, 203-211, doi: 10.1016/0042-207X(62)90978-8
CrossRef Full Text | Google Scholar
Rohm, S., Hirn, U., Ganser, C., Teichert, C., and Schennach, R. (2014). Cienkie folie celulozowe jako system modelowy dla połączeń włókien papierowych. Cellulose 21, 237-249. doi: 10.1007/s10570-013-0098-x
CrossRef Full Text | Google Scholar
Somorjai, G. A. (1981). Chemia w dwóch wymiarach: Surfaces. Ithaca, NY: Cornell University Press.
Tozuka, Y., Tashiro, E., Yonemochi, E., Oguchi, T., and Yamamoto, K. (2002). Solid-state fluorescence study of naphthalene adsorptionon porous material. J. Colloid Interface Sci. 248, 239-243. doi: 10.1006/jcis.2001.8142
PubMed Abstract | CrossRef Full Text | Google Scholar
Tozuka, Y., Yonemochi, E., Oguchi, T., and Yamamoto, K. (1998). Fluorometric studies of pyrene adsorptionon porous crystalline cellulose. J Colloid Interface Sci. 205, 510-515.
PubMed Abstract | Google Scholar
Voisin, H., Bergström, L., Liu, P., and Mathew, A. P. (2017). Materiały na bazie nanocelulozy do oczyszczania wody. Nanomaterials 7:57. doi: 10.3390/nano7030057
PubMed Abstract | CrossRef Full Text | Google Scholar
Xie, Y., Hill, C. A. S., Jalaludin, Z., and Sun, D. (2011). The water vapour sorption behaviour of three celluloses: analysis using parallel exponential kinetics and interpretation using the Kelvin-Voigt viscoelastic model. Cellulose 18, 517-530. doi: 10.1007/s10570-011-9512-4
CrossRef Full Text | Google Scholar
.