クロライドは細胞外液(ECF)区画で最も豊富なアニオンである。 高クロル血症は、血漿水中の塩化物濃度の上昇と定義される。 高クロル血症と体内の相対的な塩化物過剰は、腎血流量の減少、腎臓や消化器系を含む間質性浮腫の増加、重症患者の罹患率と死亡率の上昇3、急性腎不全患者の生存率と回復率の低下6に関係しているとされています。 体内の塩化物バランスの維持に責任を負う臓器は腎臓である。 本稿では、腎臓による塩化物の取り扱いと、高クロル血症が起こりうる臨床状況について概説する。
腎臓による塩化物の取り扱い
血漿中の塩化物の濃度は、腎臓によって調節されている。 腎臓は糸球体の基底膜を越えて塩化物を自由に濾過する。 尿中に排泄される塩化物の量は、糸球体で濾過された塩化物と、ネフロンに沿って起こる一連の輸送過程によって決定される。 通常、濾過された塩化物の60%以上は近位尿細管に沿って吸収される。 近位尿細管初期では、ナトリウムはそれに比例した量の水とともに吸収されるので、ナトリウムの濃度は変化しない。 一方、重炭酸塩などの非塩素系アニオンはナトリウムとともに速やかに吸収され、濾液から除去される7 (Fig. 1A)。 近位尿細管初期セグメント(S1およびS2)でナトリウムおよび非塩化物アニオンが吸収されると、近位尿細管内腔の塩化物濃度が上昇します。 尿細管が近位尿細管の最終セグメント(S3)に到達する頃には、塩化物濃度は血漿濃度に対して高くなり、塩化物はその濃度勾配を下って受動的に吸収されるようになる(図1B)。 塩化物の経上皮透過性は重炭酸塩の透過性よりも高いので、重炭酸塩の尿細管周囲から内腔への勾配にもかかわらず、内腔から出る塩化物の輸送は尿細管液に入る重炭酸塩を上回る。

(A) 初期の近位尿細管では、ナトリウムが有機溶質、重炭酸塩、リン酸塩とともに水と等張的に吸収され、塩化物濃度が上昇する。 (B)内腔の高い塩化物濃度は、経細胞的および傍細胞的な輸送にも有利である。 近位尿細管後部の細胞間結合部は塩化物に対してより透明になり、傍細胞輸送を促進する。 内腔の重炭酸濃度が低下しても、Na+-H+交換はNaCl再吸収の役割を果たし続ける。 細胞外への塩化ナトリウムの吸収は、Na+-H+交換と塩化物-有機アニオン(ギ酸、シュウ酸)交換のカップリングによって起こることがある。 有機酸(ギ酸またはシュウ酸)は細胞内にリサイクルされます。
近位尿細管の初期には、頂膜の塩化物-アニオン(ギ酸、シュウ酸、塩基)交換体を介して塩化物の吸収も起こり、基底側膜トランスポーターを介して細胞の外に出る8(Fig.1B). 塩化カリウムや塩化アンモニウムの負荷による高クロレム代謝性アシドーシスでは、近位尿細管での塩化物再吸収が減少するが、これは塩化ナトリウム輸送を促進する有機アニオントランスポーターの減少9と、塩化物の内腔-管腔間勾配の減少が一因である
ヘンレループの太上行肢(TALH)は塩化物の再吸収に重要な部位である10。 この部位では、ナトリウム、カリウム、塩化物がナトリウム-カリウム-2塩化物共輸送体(NKCC2)を介して同時に輸送されます(図2)。 塩化物はTALH細胞に入り、電気陰性塩化物チャネルを経由するか、電気中性の塩化カリウム共輸送体を経由して、その基底側面の外に出る。 基底側塩化物チャネル(CLC-NKB)を介した塩化物の移動は、経上皮の正(内腔)-負(基底側)電位勾配の発生に寄与している。 塩化物が細胞外に移動することによって生じる細胞内正電位は、基底側電気泳動Na+-K+ ATPaseによって、3-2の割合でカリウムを細胞内に取り込み、ナトリウムを細胞外に輸送することによって相殺される。 アピカルTALH細胞膜上のROMKカリウムチャネルは、細胞から内腔へのカリウムイオンの導電性移動を通して内腔正位(細胞内負位)に寄与している。 全体として、塩化物、ナトリウム、カリウムはNKCC2を介して細胞内に入り、大部分は塩化物は基底側ClC-NKB塩化物チャネルを介して細胞外に、ナトリウムはNa+-K+ ATPaseを介して細胞外に、カリウムはROMKチャネルを介して内腔にリサイクルされるかKClコトランスポーターを介して基底側に出て行くということである。 TALHにおけるナトリウムと塩化物の輸送の緊密な結合は、バーター症候群の1つの型で、基底側塩化物チャネルの欠陥が塩化ナトリウムの再吸収を妨げ、NKCC2タンパク質の異常で観察される腎障害を模倣することで強調される。 TAL細胞周囲のKCl共輸送体などの他の輸送体はナトリウム非依存的に塩化物を輸送するが、TALHで吸収される塩化物のほとんどはナトリウム再吸収と連関している。 したがって、このセグメントのナトリウム再吸収を増加させる因子は、塩化物再吸収も増加させる。
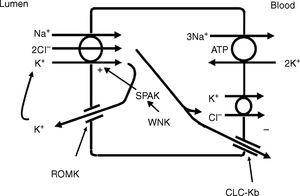
ヘンレループの太い上行肢は頂膜のNa+-K+-2Cl-共輸送体(NKCC2)を介して塩化物を吸収し、細胞から出る塩化物は基底側の塩化物チャネルとK+-Cl-共輸送によって吸収される。 K+の内腔への再循環とCLC-Kbを介した基底側伝導性塩素の排出は、内腔から基底側への正負の経上皮勾配に寄与している。 細胞内の塩化物は、細胞内のCl-が低いときにSTE20/SPS1関連プロリン/アラニンリッチキナーゼ(SPAK)とNKCC2を活性化できる塩化物感知WNKキナーゼ(WNK)を介してNKCC2の輸送を制御していると思われる。 一方、基底側クロライドチャネル出口経路の欠陥により細胞内にクロライドが蓄積すると、NKCC2の輸送が阻害される。 抗利尿ホルモンなどでNKCC2が刺激されると、塩化物の流入は増加するが、基底側Cl-伝導も亢進する。
遠位尿細管ではナトリウムと塩化物はナトリウム・クロライド共輸送体(NCC)11によって内腔から細胞内に輸送されている(図3)。 内腔から細胞内への塩化物の移動の駆動力は、基底側Na+-K+ ATPaseによって生じる内腔-細胞間ナトリウム勾配であり、これにより細胞内ナトリウム濃度を低く保つことができる。 NCCとNKCCのさらなる制御は、塩化物センサーとして機能するWNKキナーゼを介して行われると考えられ12、輸送またはそのリン酸化状態を変更することによってこれらのトランスポーターを制御できる13。遠位輸管の後半部では、頂膜上皮ナトリウムチャネル(ENaC)によるナトリウム移動によって生じる負の内腔電力も受動的塩化物再吸収の駆動力として機能しうる。
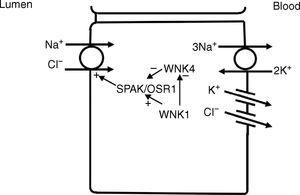
遠位尿細管では、内腔のナトリウムと塩化物はNa+-Cl-共輸送体(NCC)を介して細胞内に取り込まれる。 NCCを介した輸送は、主に基底側Na+-K+ ATPaseによって生成される低い細胞内ナトリウムによって駆動される。 WNK1キナーゼはWNK4キナーゼによるNCCの阻害をブロックする塩化物センサーとして機能すると考えられる。
集合管は最終尿中の塩化物含有量を決定する上で重要な役割を果たす. ネフロンのこの部分における塩化物の再吸収は、低塩化物摂取に対応して塩化物を保存するのに役立ち、高塩化ナトリウム食の高血圧効果に寄与しうる。 集合管で再吸収されるナトリウムのほとんどは、アルドステロンが制御する頂膜上皮ナトリウムチャネルを介して主細胞で発生する。 集合管における塩化物再吸収は、ENaCを介した内腔-細胞間ナトリウム流によって生じる内腔負の経上皮電位によって駆動される傍細胞性塩化物吸収を介して起こることがある(図4A)。 また、B型および非A非B型間充織細胞では、塩化物-重炭酸交換体であるペンドリンを介して、塩化物が内腔から細胞へ移動し、重炭酸が内腔に分泌されることにより、塩化物を輸送することができる(Fig. 4B)。 ネフロンのこの部分における様々なナトリウムと塩化物の輸送プロセスの関係は、Valletらによる最近の論文で示されている14。著者らは、腎臓におけるENaCとペンドリンのタンパク質レベルへの影響を調べるために、多くの生理学的操作を行った。 長期のNaCl負荷は、ペンドリンタンパク質レベルを有意に減少させる一方で、「活性」ENaC-γサブユニットの減少およびβサブユニットの増加が見られた。 ナトリウムと塩化物の輸送の解離が観察されたが、ヒドロクロロチアジドによるナトリウム-塩化物共輸送体の阻害で、ペンドリンレベルは低下し、ENaCレベルは増加した。 ペンドリンによる重炭酸塩分泌により内腔の重炭酸塩濃度が上昇すると、下流のENaCの活性が上昇し、ナトリウムの再吸収に影響を及ぼすと考えられる16。 層間膜細胞による塩化ナトリウム輸送は、チアジド感受性の頂膜ナトリウム依存性塩化重炭酸交換体 (NDCBE, Slc4A8) が存在することによっても促進され、1個の塩化物イオンと2個の重炭酸イオンが細胞から出る代わりに内腔から細胞に輸送されるという。 NDCBEの輸送がペンドリンを介した塩化物-炭酸水素交換と結合した場合、ナトリウムと塩化物が細胞に入る一方で炭酸水素が細胞内外を循環するため、2つの輸送体が一緒に働くと、内腔から塩化ナトリウムが正しく再吸収されると考えられる17 (図4B). この2つの陰イオン交換体の量または活性の比率を変える要因が、重炭酸塩の分泌と塩化物の再吸収に与える正味の影響を決定すると思われる。 体内の過剰な塩化物の排泄に関与していると思われるもう一つのトランスポーターはSlc26A9トランスポーターで、集合管の髄質部分において塩化物チャネルとして機能していると思われる18。これは塩化物過剰の条件下で塩化物分泌を増加させることにより、塩化物負荷の影響を修正する可能性がある。 この遺伝子をノックアウトすると、高血圧の素因となる。 Slc26a9トランスポーターは大きな塩化ナトリウム負荷の処理に重要な役割を果たすと思われるが、様々な塩化ナトリウム負荷に対応した本来のトランスポーター活性の制御はまだ不明である。 塩化物の吸収の一部は内腔の負電位と傍細胞運動によって駆動される。 (B)塩化物の細胞内再吸収は、頂膜のペンドリン塩化物-重炭酸交換体とSLCA48ナトリウム依存塩化物-2重炭酸交換体(NDCBE)の結合によっても起こりうる。 NDCBEは1個の塩化物と2個の重炭酸塩を輸送し、1個の塩化物を細胞外に排出する。 その結果、1つのナトリウムと1つの塩化物が細胞内に輸送されることになる。 アピカルペンドリンとNDCBEの活性の違いは、Cl-の分泌が優勢か吸収が優勢かを決定することができた。
(A)集合管で塩化物は分泌されるか再吸収される。 塩化物吸収の一部は内腔の負電位と傍細胞運動によって駆動される。 (B)塩化物の細胞内再吸収は、頂膜のペンドリン塩化物-重炭酸交換体とSLCA48ナトリウム依存塩化物-2重炭酸交換体(NDCBE)の結合によっても起こりうる。 NDCBEは1個の塩化物と交換に1個のナトリウムと2個の重炭酸塩を輸送し、ペンドリンの2サイクルは2個の塩化物を重炭酸塩と交換して細胞内に入れることになる。 正味のところ、1ナトリウムと1クロライドが細胞内に輸送されることになる。 アピカルペンドリンとNDCBEの活性の違いにより、Cl-の分泌が優勢か吸収が優勢かが決まる可能性があります。
クロライド濃度と高クロレミア
血清クロライド値は一般的には血清1容量中の塩素濃度として測定されています。 生物学的に活性な塩化物濃度は、血漿水中の遊離塩化物の濃度である。 19 多くの検査室で見られる自動化された方法では、血清試料を試薬で希釈して、試料の体積が通常の水分含有量であると仮定し、通常の希釈倍率を仮定して推定している。 高トリグリセリド血症や多発性骨髄腫のように、血清中の固形成分が非常に多い場合、偽性低血糖が起こることがある。 偽性低クロル血症は、臭化物またはヨウ化物の中毒でも見られることがある。 20,21
真の高クロル血症の原因水分喪失による高クロル血症
高クロル血症には、いくつかのメカニズムがある(表1)。 22 脱水状態では、腎臓は水を節約し、尿量を減少させようとする反応を示す。 脱水の程度が重くなると、体積減少の要素も加わるため、近位尿細管での塩化物およびその他の溶質の再吸収が増加し、より遠位のネフロンセグメントへの塩化物およびナトリウムの供給が減少することにより、塩化物とナトリウムの保存が行われる。 尿細管液とその内容物の近位尿細管再吸収の亢進は、液の吸収が等位的に起こるため、必ずしも塩化物濃度を変化させない。 水不足の治療は、ナトリウムと塩化物濃度の両方を低下させる電解質を含まない水の賢明な投与である。
高クロロ血症の原因。
偽性高クロル血症
サンプル希釈を伴う測定で血清固形分(脂質またはタンパク質)が多量に含まれる場合。 臭化物中毒またはヨウ化物中毒
過剰な塩化物投与
0.5mgの大量投与
0.5mgの大量投与
過剰な塩素投与9%(通常)塩化ナトリウム溶液の大量投与
高張食塩水の投与
塩水溺死
純水喪失
発熱
発汗
不十分な水分量
糖尿病
電解質過剰による水分喪失
特定の形態の下痢
浸透圧利尿
特定の症例の後遺症閉塞性利尿
代謝性アシドーシスに伴うもの
特定の形態の下痢
腎尿細管アシドーシス
炭酸脱水酵素阻害剤
尿管分岐(e.m.)、尿道灌流(e.m.)。g., 回腸膀胱)<9229><5566><5995>塩化アンモニウム投与<9229><5566><5995>アルギニンまたは塩酸リジン投与<9229><5566><5995>慢性腎臓病の特定のケース<9229><5566><5995>酸アニオンが急速に排泄される有機酸中毒(e.g., トルエンの過量投与)
呼吸性アルカローシス
過剰な塩素曝露による高クロル血症
体が塩素を多く含む液体にさらされると高クロル血症を起こすことがある。 その極端な例として、塩水溺死/消化不良がある。 海水(平均塩分濃度は3.5%)の突然の大量投入は、塩化ナトリウム負荷を排泄する腎臓の能力を圧倒し、高ナトリウム血症と高クロロ血症がよくみられる。23 とはいえ、塩水の過剰摂取に伴う高ナトリウム血症と高クロロ血症の一部は、下痢および尿の喪失に伴う水分喪失から生じるものである23。 塩水溺死による高クロレミア患者の治療は、患者の体積状態、進行中の体液および電解質の喪失の推定、および必要に応じての水と電解質の賢明な補充によって決まる。
塩化ナトリウム過剰負荷による高クロレミアのそれほど極端ではない例は、患者の容量蘇生に頻繁に使用する大量の等張 (0.9%) 塩化ナトリウム溶液 (通常食塩) を投与する場合です。 健常者に等張食塩水を大量に投与した場合、ナトリウムと塩化物のバランスが治療前の状態に戻るまでに2日間かかることがあるのは注目に値する。24 このような塩化物の滞留は、生理的状態よりも高いレベルの塩化物を含む生理食塩水にさらされることによって生じる。 正常な血漿中の塩化物濃度は95-110meq/Lの範囲であるが、正常な生理食塩水は154meq/Lの塩化物濃度である。 等張食塩水に対する排泄反応が比較的遅いのは、塩化物負荷が腎血流および糸球体濾過に及ぼす影響(尿細管糸球体フィードバック)に関連していると思われる。 塩化ナトリウムの負荷により、塩化物再吸収トランスポーター活性の低下が起こるが、14,25,26、これらのトランスポーターの低下の速度はよく定義されていない。
等張食塩水の投与により、塩化物濃度の上昇とともに重炭酸濃度も低下することがある。 正常な生理食塩水のような超高濃度塩素含有無塩基溶液の投与による血漿中の重炭酸塩の希釈のほかに、重炭酸塩の低下と塩素濃度の上昇には他の要因が関与している可能性がある。 27 この重炭酸塩の損失は、血清重炭酸塩濃度が低い場合でも起こり得ます。ヒトの研究では、等張食塩水投与後24時間以内に、ナトリウムとカリウムの損失が塩化物の損失を上回りました。 ナトリウムとカリウムに比べ、塩化物の排泄が少ないことから、重炭酸塩やその他の有機陰イオンなどの他の陰イオンが尿中に失われ、血清重炭酸濃度の低下に寄与している可能性が示唆されます24。塩基または塩基当量と塩化物濃度がより生理的に近い平衡電解質溶液の使用は、高クロロアシドーシス発症を予防するだけではなく、通常食塩水などの高クロロ血漿に伴う有害作用をある程度回避できると考えられます28、29。 健康なヒトに生理食塩水を投与した場合、塩基を含むバランスのとれた塩水と比較して、腎血流および皮質灌流が低下したことから30、患者の体液蘇生における生理食塩水の過剰投与に懸念が持たれています。 しかし、低クロレム性代謝性アルカローシスや脳浮腫のある患者など、特定の臨床状況では、生理食塩水の使用が好ましい場合がある。 HClが直接酸性化剤として投与されることはほとんどないが、塩化アンモニウムやリジン、アルギニンなどのカチオン性アミノ酸の代謝から生成されることがある31。 HClの生成は、H+とHCO3-の反応を引き起こし、CO2の生成とHCO3-の純喪失および塩化物濃度の上昇をもたらす。
呼吸滴定重炭酸塩はCO2として体から失われている。
したがって、1ミリ当量のHClを加えるごとに、1ミリ当量の重炭酸塩が消費され、重炭酸塩レベルが下がると同じ程度に塩素レベルが上昇するようにCO2に変換されます。
尿細管性アシドーシス(近位の2型RTAおよび遠位の1または4型RTA)は高クロレム代謝性アシドーシスになります。 近位尿細管性アシドーシス(2型)では、近位尿細管での重炭酸再吸収が障害され、このセグメントからの重炭酸の喪失が増加する。 また、重炭酸塩の抽出ができないため、管腔内の塩化物濃度の正常な上昇が妨げられ、塩化物の再吸収にも若干の障害が見られる。 しかしながら、近位部RTAでは、重炭酸塩輸送の減少が塩化物輸送の減少よりも大きいため、重炭酸塩よりも相対的に多くの塩化物が再吸収されることになる。 炭酸脱水酵素阻害を近位部RTAのモデルとして使用する場合、ナトリウム、カリウム、おそらく重炭酸塩の排泄率が著しく増加する一方で、尿中塩素排泄率の比較的小さな増加に反映されるように、塩化物再吸収は重炭酸塩再吸収よりも障害されていないようだ32。
古典的な遠位型RTA(タイプ1)またはタイプ4のRTAでは、純酸分泌の減少により、アンモニウムおよび/または滴定酸の排泄が損なわれて新しい重炭酸の腎臓での生成が妨げられる。 その結果、代謝によって生成されたHClは、重炭酸塩の生成と保存および塩化物の排泄によって補償されない重炭酸塩の低下をもたらす。 腎機能が保たれている限り、非塩素酸アニオンは全身循環中に蓄積されず、比較的正常なアニオンギャップが維持される。 実際、リンや硫黄を含むアミノ酸の代謝から生じるリン酸イオンや硫酸アニオンの腎排泄は、アシドーシスによって促進される33
高クロレミア代謝性アシドーシスのもう一つの原因は、下痢である。 消化管の多くのセグメントおよび膵臓などの関連外分泌器官では、重炭酸塩が塩化物と交換されて腸内に分泌されるので、特に分泌型の下痢では、重炭酸塩の損失が塩化物の保持に関連することがある34。
代謝性アシドーシスの高クロレミア型の修復には、重炭酸塩損失またはHCl生成の進行中の原因を停止させ、患者に重炭酸塩または塩基等価物(例えば、クエン酸)を与えるか、腎機能が比較的正常なら患者の腎臓で重炭酸塩を再生させることが含まれる。 代謝性アシドーシスの発生中、最初はナトリウムの純喪失と体積収縮がある。 アシドーシスがさらに長引くと、アルドステロンの高値と集合管でのENaCのアップレギュレーションにより、ナトリウムの貯留が起こる可能性がある。35 アシドーシスを修正するために重炭酸塩を供給すると、重炭酸塩は近位尿細管に留まり、通常の塩素再吸収も再確立される。 重炭酸塩による体積の再膨張は、塩化物の減少に寄与している可能性がある。 腎臓が代謝性アシドーシスを修復すると、塩化アンモニウムは尿中に排泄され、近位尿細管でグルタミン代謝の副産物として作られた重炭酸塩は血液に戻される<9229>キーポイント一覧<1512><9696><5995>腎臓は体内の塩素バランスを保つために重要な役割を担っている。 腎臓の塩化物輸送はナトリウム輸送と連関しているが、塩化物輸送は時にナトリウム輸送と乖離することがある。
高クロル血症は、水分枯渇、塩化物の過剰暴露、代謝性アシドーシスなど様々な状態によって生じる。
高尿酸血症の病因は、その障害をどのように治療すべきかの指針となる:水枯渇は、賢明な水の補充で治療し、過剰な塩素投与はさらなる塩素投与を保留し、高血糖代謝性アシドーシスは重炭酸を投与する:
利益相反
著者は、利益相反を宣言しない:
高尿酸血症の病因は、高血糖の原因となる水の補充や塩素投与、高血糖代謝性アシドーシスである。