InterPro contiene tre entità principali: proteine, firme (chiamate anche “metodi” o “modelli”) e voci. Le proteine in UniProtKB sono anche le entità proteiche centrali in InterPro. Le informazioni su quali firme corrispondono significativamente a queste proteine sono calcolate man mano che le sequenze sono rilasciate da UniProtKB e questi risultati sono resi disponibili al pubblico (vedi sotto). Le corrispondenze delle firme con le proteine determinano il modo in cui le firme sono integrate insieme nelle voci di InterPro: la sovrapposizione comparativa dei set di proteine abbinate e la posizione delle corrispondenze delle firme sulle sequenze sono usate come indicatori di parentela. Solo le firme ritenute di qualità sufficiente vengono integrate in InterPro. A partire dalla versione 81.0 (rilasciata il 21 agosto 2020) le voci di InterPro hanno annotato il 73,9% dei residui trovati in UniProtKB con un altro 9,2% annotato da firme che sono in attesa di integrazione.

InterPro include anche i dati per le varianti di splice e le proteine contenute nei database UniParc e UniMES.
Database membri del consorzio InterProModifica
Le firme di InterPro provengono da 13 “database membri”, che sono elencati di seguito.
CATH-Gene3D Descrive famiglie di proteine e architetture di dominio in genomi completi. Le famiglie di proteine sono formate usando un algoritmo di clustering di Markov, seguito da un clustering multi-linkage secondo l’identità di sequenza. La mappatura della struttura prevista e dei domini di sequenza è intrapresa utilizzando librerie di modelli di Markov nascosti che rappresentano i domini CATH e Pfam. L’annotazione funzionale è fornita alle proteine da più risorse. La predizione funzionale e l’analisi delle architetture di dominio sono disponibili sul sito web di Gene3D. CDD Conserved Domain Database è una risorsa di annotazione delle proteine che consiste in una collezione di modelli di allineamento di sequenze multiple annotate per antichi domini e proteine a lunghezza intera. Questi sono disponibili come matrici di punteggio specifico della posizione (PSSMs) per l’identificazione rapida dei domini conservati nelle sequenze proteiche tramite RPS-BLAST. HAMAP sta per High-quality Automated and Manual Annotation of microbial Proteomes. I profili HAMAP sono creati manualmente da curatori esperti e identificano le proteine che fanno parte di famiglie o sottofamiglie di proteine ben conservate codificate da batteri, archei e plastidi (cioè cloroplasti, cianelle, apicoplasti, plastidi non fotosintetici). MobiDB MobiDB è un database che annota il disordine intrinseco nelle proteine. PANTHER PANTHER è una grande collezione di famiglie di proteine che sono state suddivise in sottofamiglie funzionalmente correlate, utilizzando la competenza umana. Queste sottofamiglie modellano la divergenza di funzioni specifiche all’interno delle famiglie di proteine, permettendo un’associazione più accurata con la funzione (classificazioni di funzioni molecolari e processi biologici curate dall’uomo e diagrammi di percorsi), così come l’inferenza degli aminoacidi importanti per la specificità funzionale. Modelli di Markov nascosti (HMM) sono costruiti per ogni famiglia e sottofamiglia per classificare ulteriori sequenze di proteine. Pfam è una grande collezione di allineamenti di sequenze multiple e modelli di Markov nascosti che coprono molti domini e famiglie di proteine comuni.
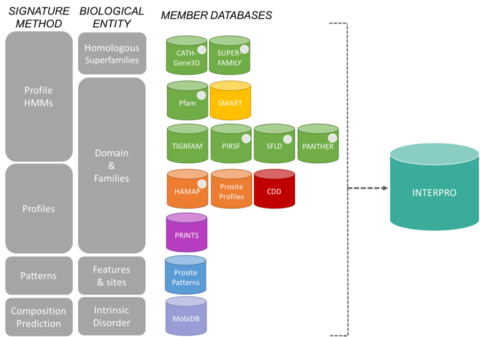
Il sistema di classificazione delle proteine PIRSF è una rete con livelli multipli di diversità di sequenza dalle superfamiglie alle sottofamiglie che riflette la relazione evolutiva delle proteine a lunghezza intera e dei domini. L’unità primaria di classificazione PIRSF è la famiglia omeomorfa, i cui membri sono sia omologhi (evoluti da un antenato comune) che omeomorfi (che condividono la somiglianza della sequenza full-length e un’architettura di dominio comune). PRINTS PRINTS è un compendio di impronte digitali di proteine. Un’impronta digitale è un gruppo di motivi conservati usati per caratterizzare una famiglia di proteine; il suo potere diagnostico è raffinato dalla scansione iterativa di UniProt. Di solito i motivi non si sovrappongono, ma sono separati lungo una sequenza, anche se possono essere contigui nello spazio 3D. Le impronte digitali possono codificare le pieghe e le funzionalità delle proteine in modo più flessibile e potente di quanto possano fare i singoli motivi, poiché la loro piena potenza diagnostica deriva dal contesto reciproco offerto dai vicini del motivo. PROSITE PROSITE è un database di famiglie e domini proteici. Consiste di siti biologicamente significativi, modelli e profili che aiutano a identificare in modo affidabile a quale famiglia proteica nota (se esiste) appartiene una nuova sequenza. SMART Simple Modular Architecture Research Tool Permette l’identificazione e l’annotazione di domini geneticamente mobili e l’analisi delle architetture di dominio. Sono rilevabili più di 800 famiglie di domini trovati in proteine di segnalazione, extracellulari e associate alla cromatina. Questi domini sono ampiamente annotati rispetto alle distribuzioni fittizie, alla classe funzionale, alle strutture terziarie e ai residui funzionalmente importanti. SUPERFAMILY SUPERFAMILY è una libreria di modelli di Markov nascosti di profilo che rappresentano tutte le proteine di struttura nota. La libreria è basata sulla classificazione SCOP delle proteine: ogni modello corrisponde a un dominio SCOP e mira a rappresentare l’intera superfamiglia SCOP a cui il dominio appartiene. SUPERFAMILY è stata utilizzata per effettuare assegnazioni strutturali a tutti i genomi completamente sequenziati. SFLD Una classificazione gerarchica degli enzimi che mette in relazione specifiche caratteristiche della struttura della sequenza con specifiche capacità chimiche. TIGRFAMs TIGRFAMs è una collezione di famiglie di proteine, con allineamenti multipli di sequenza curati, modelli di Markov nascosti (HMM) e annotazioni, che fornisce uno strumento per identificare proteine funzionalmente correlate basate sull’omologia di sequenza. Le voci che sono “equivalenti” raggruppano proteine omologhe che sono conservate per quanto riguarda la funzione.
Tipi di datiModifica
InterPro consiste di sette tipi di dati forniti da diversi membri del consorzio:
| Tipo di dati | Descrizione | Basi di dati contribuenti |
|---|---|---|
| Voci InterPro | Strutturali e/o domini funzionali di proteine predette usando una o più firme | Tutti i 13 database membri |
| Firme dei database membri | Signature dei database membri. Queste includono le firme che sono integrate in InterPro, e quelle che non lo sono | Tutti i 13 database membri |
| Proteina | Seguenze di proteine | UniProtKB (Swiss-Prot e TrEMBL) |
| Proteoma | Raccolta di proteine che appartengono ad un singolo organismo | UniProtKB |
| Struttura | 3-strutture dimensionali delle proteine | PDBe |
| Tassonomia | Informazioni tassonomiche sulle proteine | UniProtKB |
| Set | Gruppi di famiglie evolutivamente correlate | Pfam, CDD |

Tipi di voci InterProModifica
Le voci InterPro possono essere ulteriormente suddivise in cinque tipi:
- Superfamiglia omologa: Un gruppo di proteine che condividono una comune origine evolutiva come si vede nelle loro somiglianze strutturali, anche se le loro sequenze non sono altamente simili. Queste voci sono specificamente fornite solo da due database membri: CATH-Gene3D e SUPERFAMILY.
- Famiglia: Un gruppo di proteine che hanno una comune origine evolutiva determinata attraverso somiglianze strutturali, funzioni correlate, o omologia di sequenza.
- Dominio: Un’unità distinta in una proteina con una particolare funzione, struttura o sequenza.
- Ripetizione: Una sequenza di aminoacidi, solitamente non più lunga di 50 aminoacidi, che tende a ripetersi molte volte in una proteina.
- Sito: Una breve sequenza di aminoacidi dove almeno un aminoacido è conservato. Questi includono siti di modifica post-traslazione, siti conservati, siti di legame e siti attivi.