- Contenuti
- Funzione
- Meccanismo di inibizione dell’actio strand-transfer
- HIV e AIDS
- Impatto della struttura
- PFV Intasome Crystallization
- Tecnica di cristallizzazione
- Statistiche cristallografiche e di raffinamento
- Architettura complessiva & Componenti
- Struttura
- Integrasi e interazioni con il DNA
- Sito attivo
- Inibitori dell’integrasi
Contenuti
- 1 Funzione
- 2 Meccanismo per l’inibizione del trasferimento di filamenti
- 3 HIV e AIDS
- 4 Impatto della struttura
- 5 PFV Intasoma Crasometransfer inhibition actio
- 3 HIV e AIDS
- 4 Impatto della struttura
- 5 Cristallizzazione di PFV Intasome
- 5.1 Tecnica di cristallizzazione
- 5.2 Statistiche cristallografiche e di raffinamento
- 6 Architettura complessiva & Componenti
- 6.1 Struttura
- 6.2 Interazioni tra Integrasi e DNA
- 6.3 Sito attivo
- 7 Inibitori dell’Integrasi
- 7.1 Risorse aggiuntive
- 8 Strutture 3D dell’integrasi retrovirale
Funzione
L’integrasi retrovirale è un enzima retrovirale essenziale che si lega al DNA virale e lo inserisce nel cromosoma della cellula ospite. Il cDNA trascritto al contrario del virus dell’immunodeficienza umana di tipo 1 (HIV-1) viene inserito nel genoma della cellula ospite per aumentare la fitness e la virulenza del patogeno. L’integrasi è prodotta da una classe di retrovirus (come l’HIV) ed è usata dal virus per incorporare il suo materiale genetico nel DNA della cellula ospite. Il macchinario cellulare dell’ospite produce quindi mRNA e poi proteine dal materiale genetico incorporato, replicando così il virus. Sebbene siano stati studiati diversi farmaci che inibiscono l’integrasi, il meccanismo responsabile dell’azione di inibizione del trasferimento dei filamenti deve ancora essere chiarito. Tuttavia, Hare el al (2010) hanno determinato i costituenti strutturali dell’integrazione retrovirale. Un’ulteriore delucidazione della struttura completa dell’integrasi retrovirale e la sua applicazione per regolare le attività funzionali ed enzimatiche potrebbe potenzialmente permettere ai ricercatori di ritardare la progressione delle malattie retrovirali. Inoltre, lo studio dell’integrazione di HIV-1 potrebbe portare a un nuovo bersaglio promettente e contribuire alla generazione di modelli di farmacoforo per la terapia antivirale.
Inibitori dell’integrasi dell’HIV: Raltegravir, commercializzato come Isentress è attualmente approvato come inibitore terapeutico dell’integrasi dell’HIV. È stato approvato il 12 ottobre 2007. Per l’integrasi fagica vedi Integrasi fagica.
Meccanismo di inibizione dell’actio strand-transfer
Caption
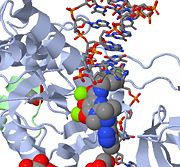
Sopra è un’immagine JMOL di MK-0518 che blocca l’estremità 3′ del DNA virale dal legare il sito attivo. Notare gli atomi di ossigeno chelanti del metallo in MK-0518 che interagiscono con i cationi di magnesio presenti nel sito attivo.
L’integrasi dell’HIV di tipo 1 è un obiettivo relativamente nuovo e inedito per gli inibitori. Nel 2007, il primo inibitore dell’integrasi HIV-1, Raltegravir, è stato approvato dalla FDA per l’uso nell’HIV-1 come terapia combinata. Gli inibitori dello strand-transfer funzionano impedendo l’integrazione concertata del DNA virale nel cromosoma dell’ospite. Dopo l’ingresso del virus nella cellula ospite, la trascrittasi inversa converte il ssRNA virale in dsDNA. A questo punto, l’integrasi forma un complesso con il DNA virale, creando il complesso di pre-integrazione (intasoma). Il complesso di pre-integrazione viene poi trasportato nel nucleo, dove due nucleotidi vengono rimossi dall’estremità 3′. Successivamente, il DNA viene integrato covalentemente nel DNA dell’ospite. Gli inibitori dello strand-transfer interrompono questo processo, impedendo l’integrazione del DNA virale nel cromosoma dell’ospite. Gli inibitori dello strand-transfer funzionano impegnando i cofattori degli ioni metallici che si trovano nel sito attivo dell’integrasi retrovirale. Gli atomi di ossigeno chelanti trovati negli inibitori interagiscono direttamente con i cofattori metallici, mentre il gruppo alobenzilico si inserisce nella tasca creata dal 3′ DNA virale spostato nel sito attivo.
HIV e AIDS

Nel 2010, ci sono più di 25 milioni di persone sono morte di AIDS, e si stima che circa 33 milioni di persone vivono con l’HIV.
Ad oggi, più di 25 milioni di persone sono morte di AIDS e si stima che circa 33 milioni di persone vivono con l’HIV oggi. Per gli inibitori dell’integrasi retrovirale vedi Raltegravir e Farmacocinetica degli inibitori dell’integrasi retrovirale.
Impatto della struttura
Le strutture tridimensionali di alcune proteine della cellula ospite critiche per comprendere il meccanismo dell’infezione da HIV e la virulenza sono emerse dalle analisi cristallografiche a raggi X. Le strutture della proteasi e dell’integrasi dell’HIV sono tra le strutture di più alto livello che hanno contribuito a salvare molte vite e ad aumentare la qualità della vita di molti individui affetti da HIV. Viene implementata nella progettazione di farmaci basata sulla struttura per sviluppare inibitori della proteasi e dell’integrasi, ed è usata come componente significativa della terapia antiretrovirale altamente attiva (HAART).
Mentre gli agenti antiretrovirali esistenti migliorano la qualità della vita e prolungano la vita di molti pazienti, non riescono a sradicare la malattia. Gli studi sugli inibitori dell’integrasi mostrano che la combinazione con altri farmaci antiretrovirali diminuisce gli adattamenti virali, e può avere il potenziale per essere usata per la terapia di salvataggio per i pazienti che hanno acquisito resistenza ad altri farmaci. Per saperne di più, vedi
- AIDS Before Protease Inhibitors & HIV Protease Inhibitors: A Breakthrough
- Treatments:Retroviral Integrase Inhibitor Pharmacokinetics References.
PFV Intasome Crystallization
To mimic the viral DNA ends of HIV-1, Hare et al (2010) used soluble and fully functional prototype foamy virus (PFV) intasome preparations, obtained using recombinant PFV integrase and double-stranded oligonucleotides.
La notevole stabilità dei complessi integrasi-DNA è stata determinata osservando le reazioni di strand transfer in vitro, che sono state classificate in tre modalità di migrazione della deproteina: (1) eventi concertati singoli: plasmide bersaglio linearizzato; (2) eventi concertati multipli: striscio; (3) eventi half-site: DNA circolare aperto. Un’ulteriore caratterizzazione dell’intasoma PFV ha anche mostrato una sostanzialità strutturale che implicava forti interazioni proteina-proteina e proteina-DNA nonostante l’incubazione prolungata in condizioni di alta forza ionica. Saggi completi di cristallizzazione hanno effettuato una configurazione cristallina praticabile che diffrangeva i raggi X con una risoluzione di 2,9 Angstroms. Alla fine è stata determinata una struttura tridimensionale. L’unità asimmetrica conteneva un singolo dimero integrasi con una molecola di DNA virale stabilmente legata, e una coppia di dimeri integrasi consociati con simmetria, che formavano un tetramero oblungo. L’interfaccia del dimero è stabilizzata dalle interazioni intermolecolari dei domini amino terminali e del nucleo catalitico (subunità interna- subunità esterna). La forma complessiva del tetramero oblungo è unica, sebbene presenti somiglianze con i complessi dell’integrasi HIV-1 precedentemente riportati.
Tecnica di cristallizzazione
I complessi proteina-DNA sono stati formati utilizzando la lunghezza completa, PFV IN wild-type e dsDNA sintetico che ha modellato l’estremità virale.
L’intasoma è stato cristallizzato utilizzando la tecnica di diffusione con goccia di vapore sospeso. La soluzione di riserva consisteva in solfato di ammonio 1,35 M, 25% (v/v) di glicerolo, 4,8% (v/v) di 1,6-esandiolo e 50mM di acido 2-(N-morfolino) etansolfonico (MES) a pH 6,5. I cristalli di proteina-DNA sono stati anche impregnati in presenza di MK0518, GS9137, Mg(II), e/o Mn(II). La struttura cristallina è stata risolta usando la sostituzione molecolare.
Statistiche cristallografiche e di raffinamento
Hare et al (2010) hanno pubblicato dati su sette strutture cristalline. Questi dati includono il complesso PFV IN (forma apo) e sei strutture aggiuntive, compreso il complesso legato a Mg, Mn, Mg/MK0518, Mn/MK0518, Mg/GS9137, e Mn/GS9137. Tutte e sette le strutture appartengono al gruppo spaziale P41212. Sono state raffinate ad una risoluzione compresa tra 2.85 e 3.25 Å.
Architettura complessiva & Componenti
Struttura
La struttura complessiva dell’intasoma PFV assemblato è un modello tetramerico basato su due strutture di dominio con un’interfaccia dimer-dimero. I precedenti modelli di intasoma descrivono una struttura simile ma più flessibile, mentre l’intasoma PFV ha dimostrato di essere altamente vincolato. Usando la modellazione omologica, Hare et al (2010) propongono che i linker interdominio più corti possano essere un fattore di flessibilità, in particolare nell’integrasi HIV-1. Le subunità interne del tetramero sono implicate nella tetramerizzazione complessiva e nel legame al DNA virale. I domini del nucleo catalitico delle subunità esterne possono agire come supporti, ma poiché i domini amino- e carbossi-terminali non sono risolti nelle mappe di densità elettronica, la loro funzione rimane inconcludente. Il dominio del nucleo catalitico e il linker del dominio carbossi-terminale adottano una conformazione estesa per la maggior parte della sua lunghezza, e sono situati parallelamente al dominio amino-terminale e al linker del dominio del nucleo catalitico della subunità interna. I linker interdominio I linker interdominio (CCD-CTD linker e NTD-CCD linker) legano insieme le due metà dell’intasoma, e la struttura è ulteriormente stabilizzata da una coppia di domini carbossi-terminali che interagiscono con entrambi i domini del nucleo catalitico interno.
Integrasi e interazioni con il DNA
Forte interazioni proteina-DNA si trovano nei sei nucleotidi terminali. Ogni dominio terminale carbossilico interagisce con la spina dorsale del fosfodiestere di entrambe le molecole di DNA virale. Inoltre, il dominio amino-terminale-dominio di estensione e il dominio amino terminale interagiscono con il DNA virale nel sito attivo del dominio del nucleo catalitico opposto.
Sito attivo
I carbossilati del sito attivo sono catene laterali di Asp 128, Asp 185, Glu 221.
Un atomo di zinco ciascuno è situato vicino ai siti attivi.
- .
- .
Inibitori dell’integrasi
| Nome | Marca | Azienda | Brevetto | Note |
| Raltegravir | Isentress | Merck & Co. | – | conosciuto anche come MK-0518. L’isopropilico e il metil-ossadiazolo di MK-0518 sono coinvolti in interazioni idrofobiche e di stacking con le catene laterali di Pro 214 e Tyr 212 per stabilizzare questo farmaco all’interno del sito attivo dell’intasoma PFV. Questo modo di interazione farmaco-legante provoca lo spostamento dell’estremità reattiva 3′ del DNA virale dal sito attivo dell’intasoma del PFV. Dopo il legame di MK-0518 al sito attivo, il gruppo idrossile 3′ reattivo si allontana dal sito attivo dell’intasoma del PFV per più di 6 Angstrom. Raltegravir è stato approvato dalla FDA il 12 ottobre 2007, per l’uso con altri agenti anti-HIV nel trattamento dell’infezione da HIV negli adulti. È il primo inibitore dell’integrasi approvato dalla FDA. |
| Elvitegravir | – | Gilead Science | – | GS-9137 interagisce con il Pro 214 dell’intasoma di PFV attraverso la sua base chinolonica e il gruppo isopropilico. In fasi sperimentali; condivide la struttura centrale degli antibiotici chinolonici. Gli studi di fase II di elvitegravir in persone con esperienza di trattamento sono stati completati. Sono in corso studi di fase III in pazienti con esperienza di trattamento. È in corso uno studio di fase II di elvitegravir in persone che non hanno mai assunto una terapia antiretrovirale. Questo studio valuterà anche un agente di potenziamento al posto del Norvir, attualmente chiamato GS9350. Elvitegravir è promettente per i pazienti HIV-positivi che hanno preso altri farmaci anti-HIV in passato. |
| MK-2048 | – | Merck & Co. | – | Un inibitore dell’integrasi di seconda generazione, destinato ad essere usato contro l’infezione da HIV. È superiore al primo inibitore dell’integrasi disponibile, raltegravir, in quanto inibisce l’enzima HIV integrasi 4 volte più a lungo. È in fase di studio per l’uso come parte della profilassi pre-esposizione (PrEP). |
Vedi anche Farmacocinetica degli inibitori dell’integrasi retrovirale.